People have asked why going different suggestion choices give different results – sometimes contradictory ones! The suggestions are determined by the bacteria included to alter. There is no magic way to select the bacteria. The site gives you a variety of choices/methods reflecting various requests expressed. This post attempts to explain these choices. Remember a typical microbiome result may be 600 bacteria – picking a dozen bacteria at random will give different suggestions every time. Many of the bacteria are ‘noise’ with no health impact in most cases
Quick Suggestions
This looks at only the bacteria in Dr. Jason Hawrelak criteria for a healthy gut. If you are outside of his ranges then low values are attempted to be increased and high values are decreased.
Number of Bacteria Considered: 15
If any other published author care to provide their criteria and grant permission to use, it can be added.
Medical Conditions
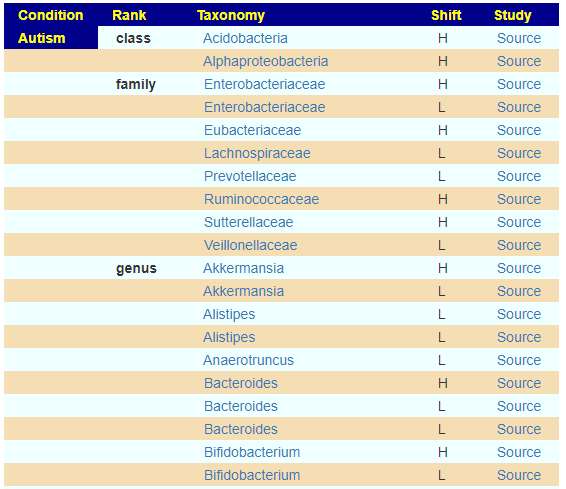
When you click on one of the items on the “Adjust Condition A Priori” link there is no microbiome to refer to so one is synthetically created. This is done by looking at the reported shifts and computing one.
We apply some fuzzy logic here.
- If just one report, we run with that value
- If equal number of high and low we ignore.
- If different number of high and low, we compute the difference, and deem the winner to be included
We then create a profile using the 12%ile value for low and 87% for high value.
The resulting synthetic microbiome is then processed using the 50%ile as our reference, scaling. The data to be processed may look like this:
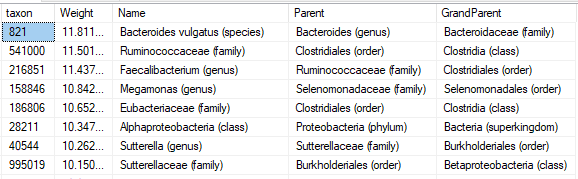
Number of Bacteria Considered: depending on condition: 5- 300 bacteria, typically 30
Advance Suggestions
This is the workhorse which gives many options to both increase and decrease bacteria included. It takes all bacteria (regardless of possible medical significance) as a starting point and adjusts them.
Add in all those I am missing that are seen in % of other samples
No bacteria is seen in every samples. Some people have none of some bacteria and they are concerned about this. This allows you to include very common bacteria that you are missing with a zero value. It is questionable if this philosophical belief have significance. The most common bacteria is listed below.
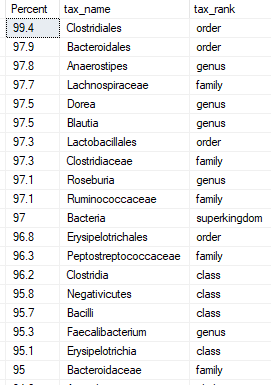
Limit to Taxonomy Rank of ….
It appears that often the real health significant items are at the lowest level of the bacteria hierarchy. There are good Lactobacillus strains and there are bad Lactobacillus strains (which have been reported to be fatal). This allows you to focus only the bottom levels. The more levels, the more bacteria are targeted – and the greater that ‘noise’ may hide what is significant.
A simple analogy. A kid at a school did some vandalism, you have a vague idea of who (the Species). Do you proceed to punish him with all of his friends (i.e. the Genus)? Do you punish those in classes that he is in (the Family) – keeping all of the classes in for detention. Do you push the entire grade in that school (the Order)? The entire school (the Class). Morale and school performance will change for those impacted.
Bacteria Selection Choices
This attempted to filter to outliers before the [My Taxa View] was created which allowed hand selection. The philosophical reasoning is that very high and very low are the most probable cause of health issues. This discards bacteria that are in the middle range. You specify if you want to focus only on:
- top/bottom 6% – Example Count: 6
- top/bottom 12% – Example Count: 35
- top/bottom 18% – Example Count: 66
Filter by High Lactic Acid/Lactate Producers
This was a special early request from a reader. It will filters to those bacteria that are lactic acid producers where the values are above the 50%ile. Everything else is excluded. This functionality has been improved using EndProducts Explorer and hand picking the taxa (thus you can do it for any end product in our system). Values are scaled from the difference to the median value.
This was retained because lactic acid issues often result in cognitive impairment, hence a simple route for those people.
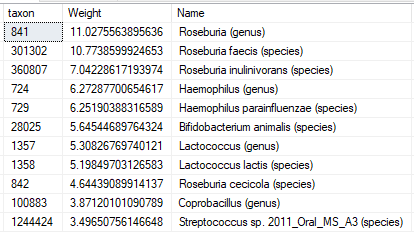
Deprecated: Filtering by….
Filtering by medical conditions, symptoms have been deprecated and replaced by hand picked taxa. This allows unlimited combinations of conditions and symptoms to be handled.

My Biome View
This is for people that wishes to ‘eye-ball’ the choice of bacteria. This shows the relative ranking/percentile and how many samples have it. For a bacteria that is seen in only a dozen sample (like Legionellaceae below) or with a count of 100 or less, is unlikely to have any significance.
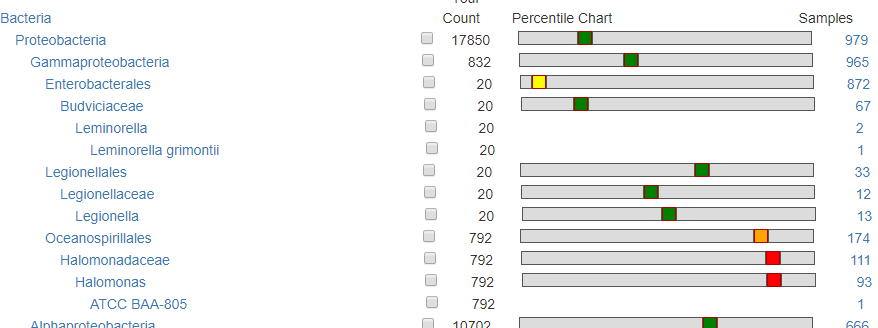
How are Hand Pick Taxa Handled?
We maintain the same pattern: What is the difference from the median/50%ile (NEVER the average) and we then scale it and feed those values into the suggestion engine.
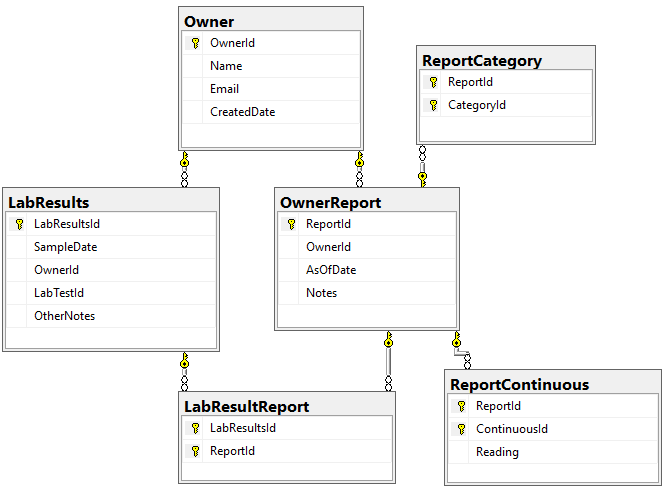
How are Lab Results handled
The original approach of giving every bacteria equal weight has been updated recently. Like with Medical Conditions above, we create a synthetic microbiome using
- 1 down arrow for 18%ile value
- 2 down arrow for 12%ile value
- 2 down arrow for 6%ile value
- 1 up arrow for 82%ile value
- 2 up arrow for 88%ile value
- 3 down arrow for 94%ile value
Bottom Line
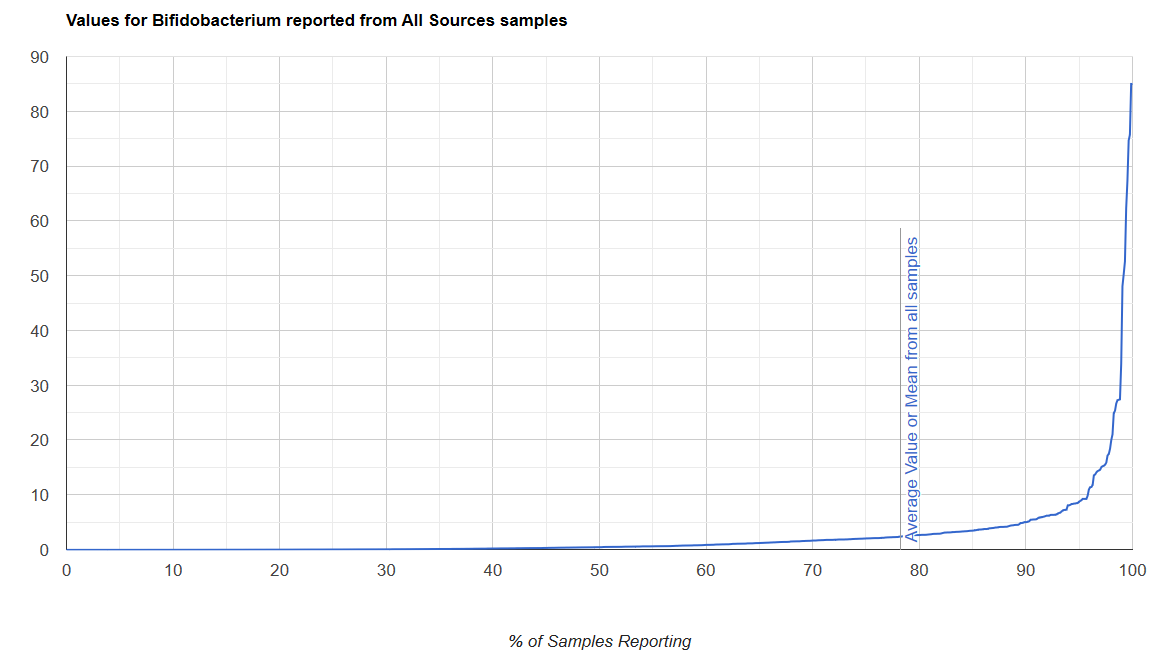
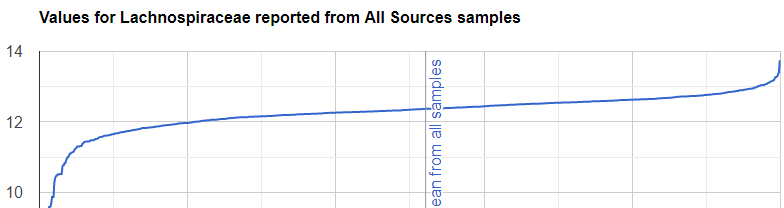
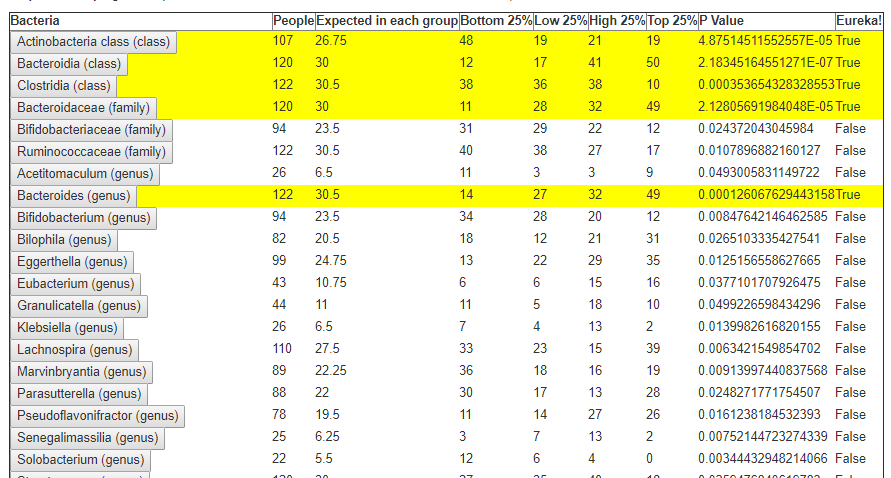
We always use a provided range (Jason Hawrelak) or the difference from the 50%ile/median. We never use Average — and feel that any lab that reference averages do not really understand the data and lack adequate statistical staff. Once upon a time, in the early days we used average but as we got familiar with the data we realized how wrong that approach was — the data is not a bell curve/normal distribution. A simple example is below.
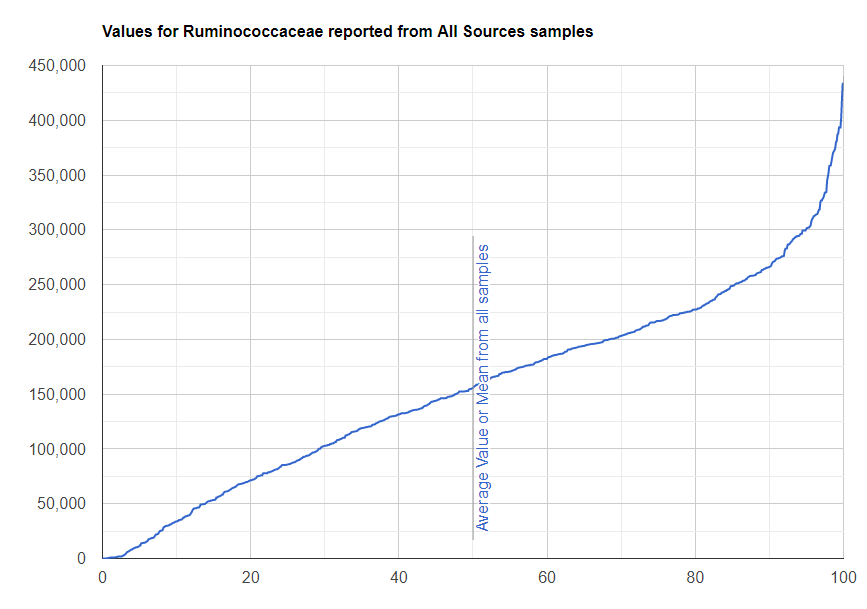
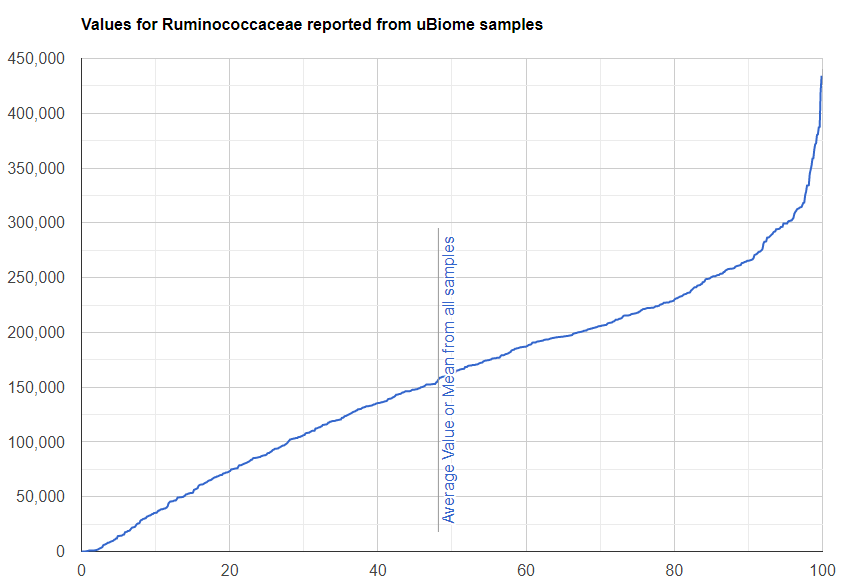
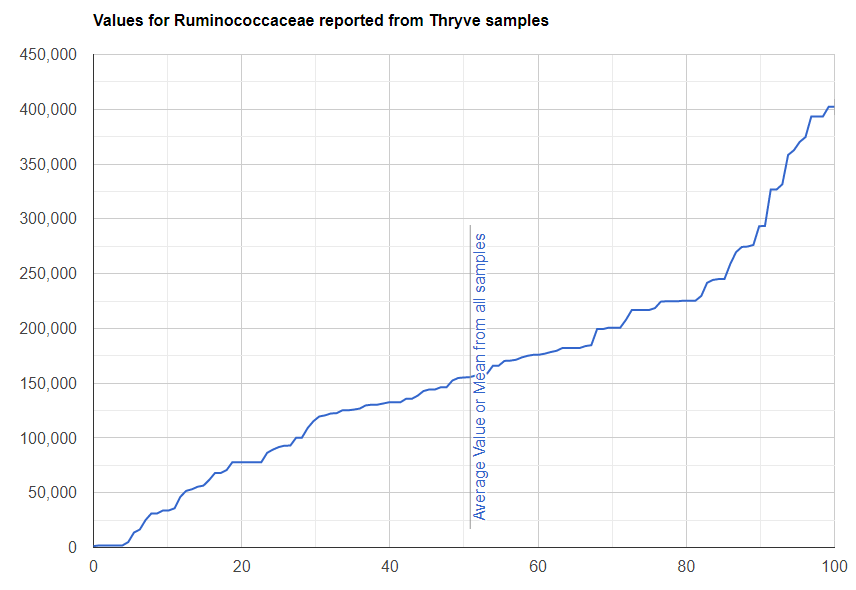
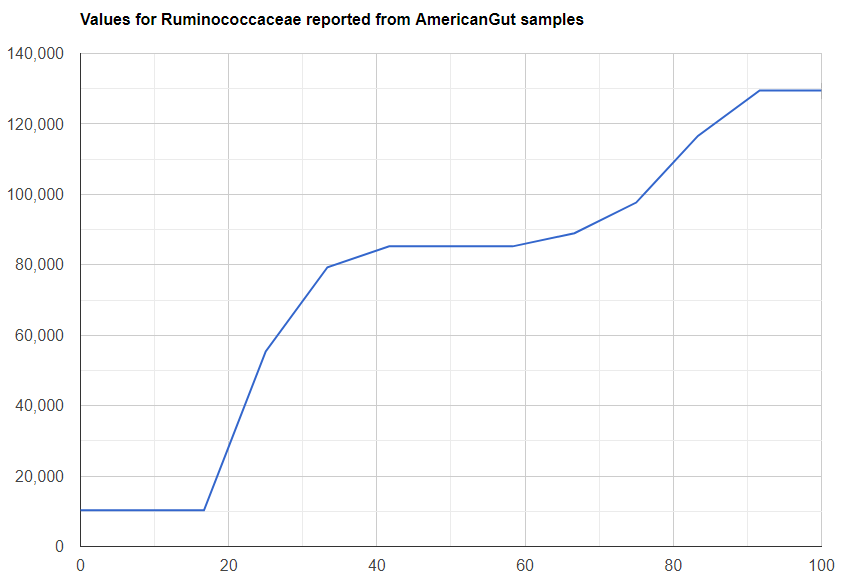
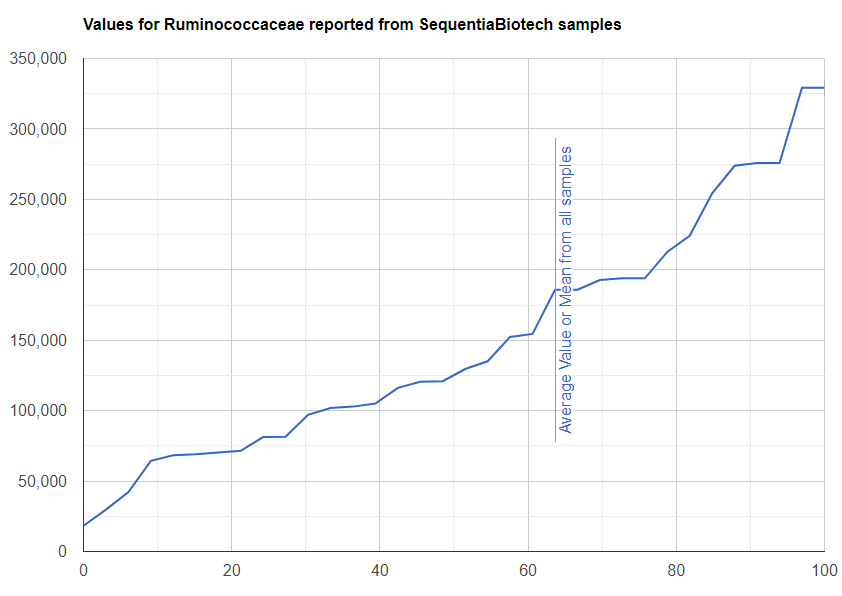


Recent Comments