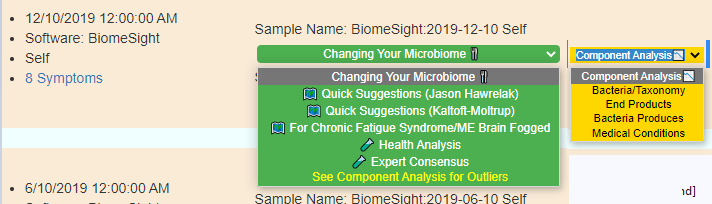
Often I am faced with requests to keep things simpler of the Microbiome Prescription site while getting requests to give more choices (often arising from people’s belief of what may work). I have just finished a revision attempting to balance these two (and set up infrastructure to give more choices in the future)
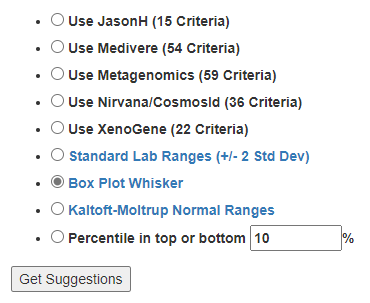
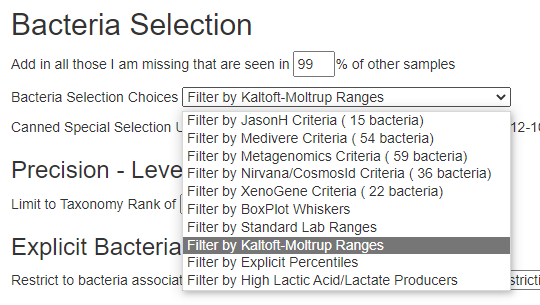
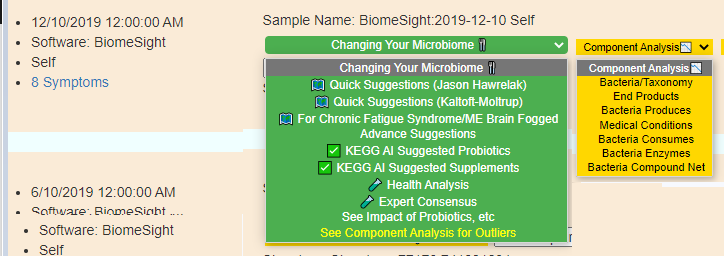
Old version menu of choices – with intermediate displayRevised version menu of choices – with intermediate display
The Expert Criteria takes you to a new page that has many criteria listed:
Expert Criteria Choices (more may be added as data becomes available)
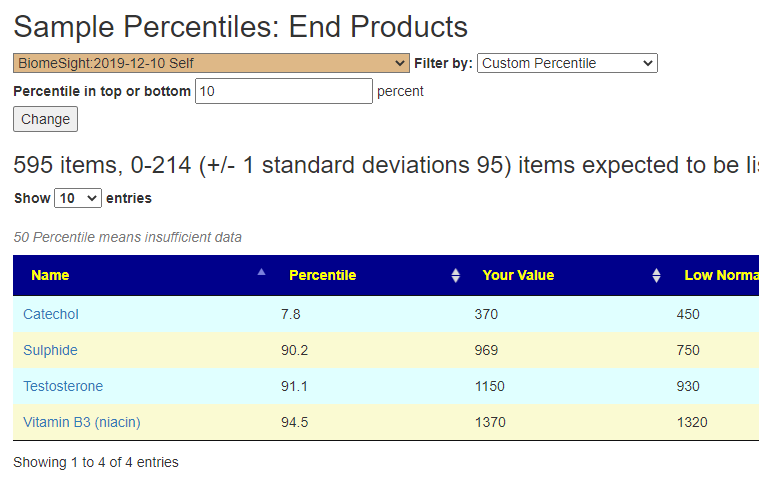
There will be differences from each choice, because the bacteria selected will likely be different. For one sample:
Why the differences? With only a few dozen bacteria with ranges, the chance of being picked is low. Box Plot and KM are computed for almost every bacteria. So with 700 bacteria, we have around 10% picked. With just 50 bacteria to be examined, we have around 14% picked.
These choices are also available in Advance Suggestions
Can you guys tell us beginners what do you guys do for gut health
If in unhealth…
My approach is simple, get a 16s (Biomesight or Ombre) microbiome test. Transfer or upload the data to https://microbiomeprescription.com/, get suggestions and do them for 4-8 weeks. Retest and repeat.
Here is an example of my applying this method (with 8 blog posts)
A key item is to rotate, rotate, rotate. Take the list of take suggestions and break into 4 groups. Do each for 2 weeks and then change to the next group. Attempt to remove ALL of the avoids (at least those with a value of 0.4 and higher). Simple enough? (Apart from the methodology to select the bacteria to alter — a new video on methodology is in progress)
If in health
Every 6-9 months, My approach is simple, get a 16s (Biomesight or Ombre) microbiome test. Transfer or upload the data to https://microbiomeprescription.com/, get suggestions. Look at rotating in (2 weeks on, 4 weeks off) any items over 0.8 on the take. Try to reduce any items on the avoid over 0.4.
If I am prescribe an ongoing medicine, then I will take a sample after 4 weeks and make modifications to counter any adverse effects. If the medication is in the existing database, I would check if there is an excessive shift (in terms of percentile) of the bacteria that it is known to shift.
For items like vaccinations and short term antibiotics, I will wait at least 8 weeks, preferred 12 weeks, to allow my immune system to settle down.
Bottom Line
That’s it. I do not do items exclusively from suggestions. I may take other items for diverse reasons — if they are not high in the avoid list, I just keep taking them.
Q&A
Q: Do you drink Kefir in general for gut health?
A: No, unpredictability of which bacteria are in it. I tend to keep to researched strains only. For yogurt: Activa is an example, or Yakurt probiotic drink. Custom Probiotics is a regular source.
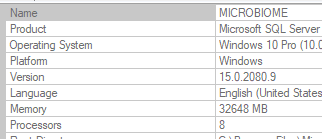
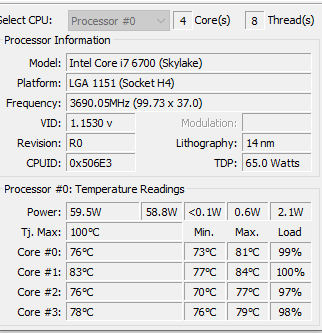
I have moved on to rework the existing code. The processing is done on a dedicated server (32GB of memory, SSD drives for SQL Server. A little over 15,700,000 combinations of taxon needs to be made. Each combination needs to get all data on these taxons that are concurrent in any samples.
I.e. All samples that have Taxon 123 and Taxon 432 being reported.
The CPU SpecificationTask Manager during the runThe Data Server Specifications
The computations are done using Parallel and Concurrent libraries. All indices have been tuned for this analysis. The SQL Server and the utility to calculate the associations are on the same server – so no latency or network impact.
I found that the CPU temperatures exceed the maximum recommended for the CPU chip.
Original Run load. The load was throttled to 85-90% to keep the temperatures lower. The log of the run is below. Elapsed time 50 hours with the computer running at 98%. This excludes time to calculate the monotonic transformation of the data (the association is neither linear nor Bayesian).
2022-03-27 20:12:08 Bacteria2Bacteria - 15,717,260 Items to Inspect
2022-03-29 22:48:34 Bacteria2Bacteria - 15,717,260 Items Inspected
2022-03-29 22:48:47 805,770 Items found
Bottom Line
This analysis likely qualifies for the “big data” label. So why is it worth it? The answer is simple, for some bacteria we have no information on what increases or decreases it. If we know which bacteria is associated with the bacteria growth or reduction, then we can synthesize modifiers of these bacteria that we lack information on.
This has not been implemented in suggestions (and will likely be available at the nerd level when it is).
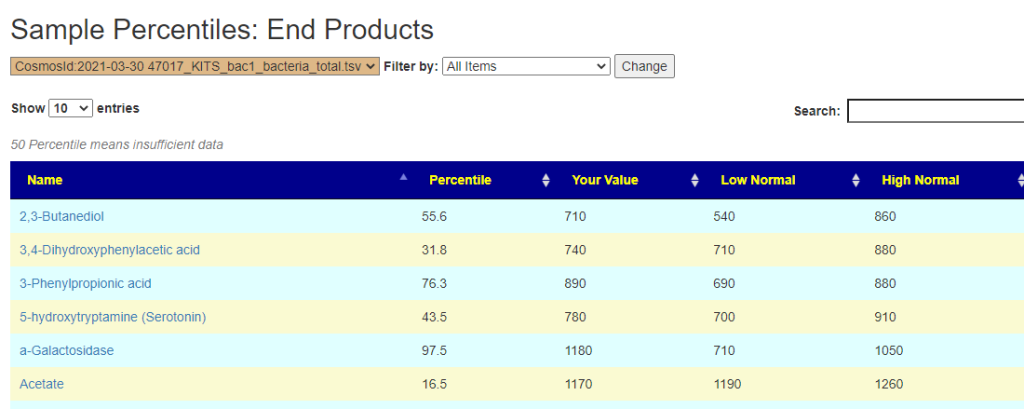
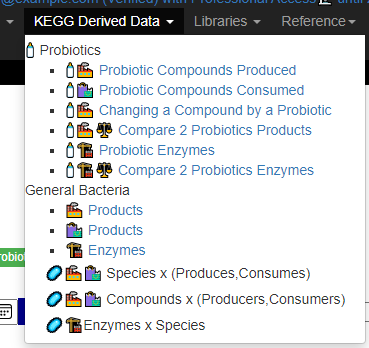
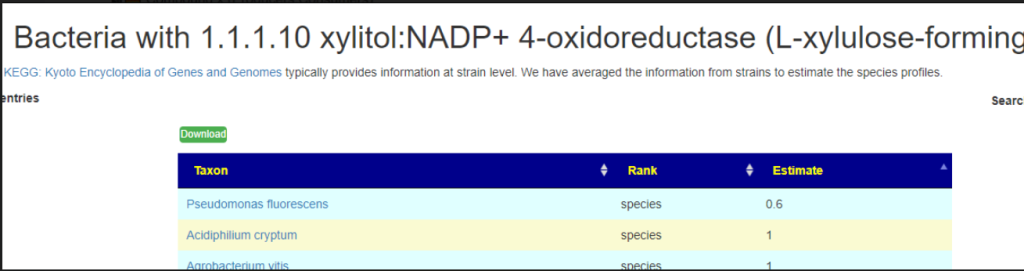
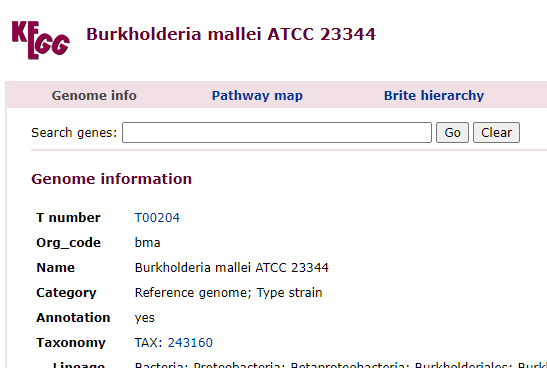
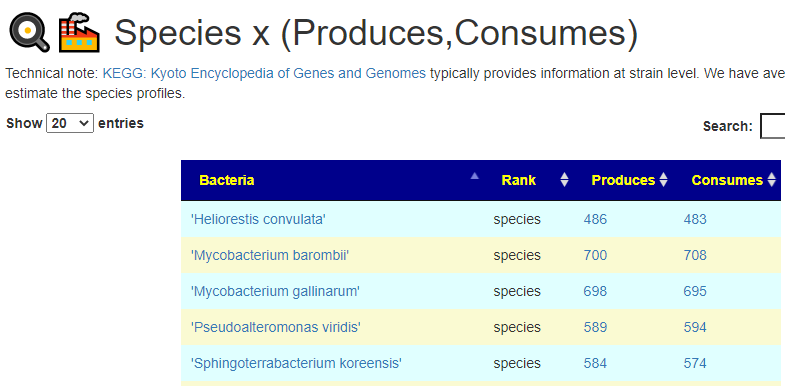
This year I have been focusing on a deep review of the code to improve accuracy and to address a variety of issues. The first item was redoing all of the KEGG Derived data (deriving compounds and enzymes at the species level instead of random strains). See KEGG Data being updated
The second item was cleaning up percentile computations on samples for:
Taxonomy
End Products
Compound Produced (KEGG)
Compound Consumed (KEGG)
Net Compound (Produced – Consumed) (KEGG)
Enzymes (KEGG)
Medical Conditions (US National Library of Medicine)
Combined with this was also implementing Display Levels across menu. See Display Levels.
Existing Menu Items
Original (overwhelming) menu
New Menu Items
Display Level: Public
Display Level: Beginner
Display Level: Intermediate
Display Level: Advance
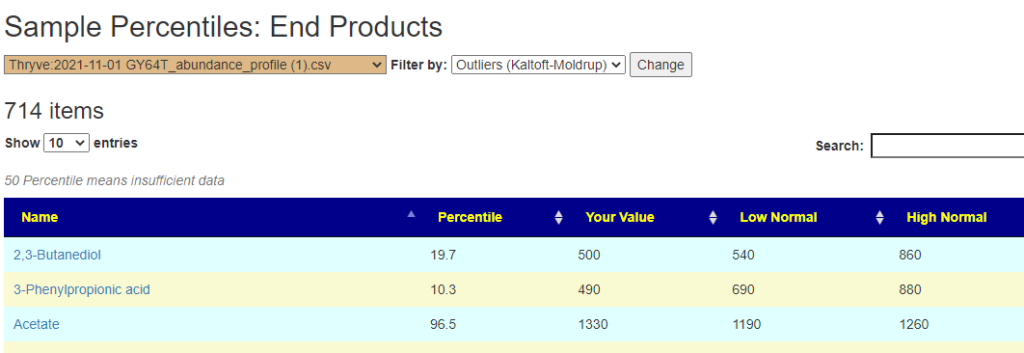
Outliers Improved
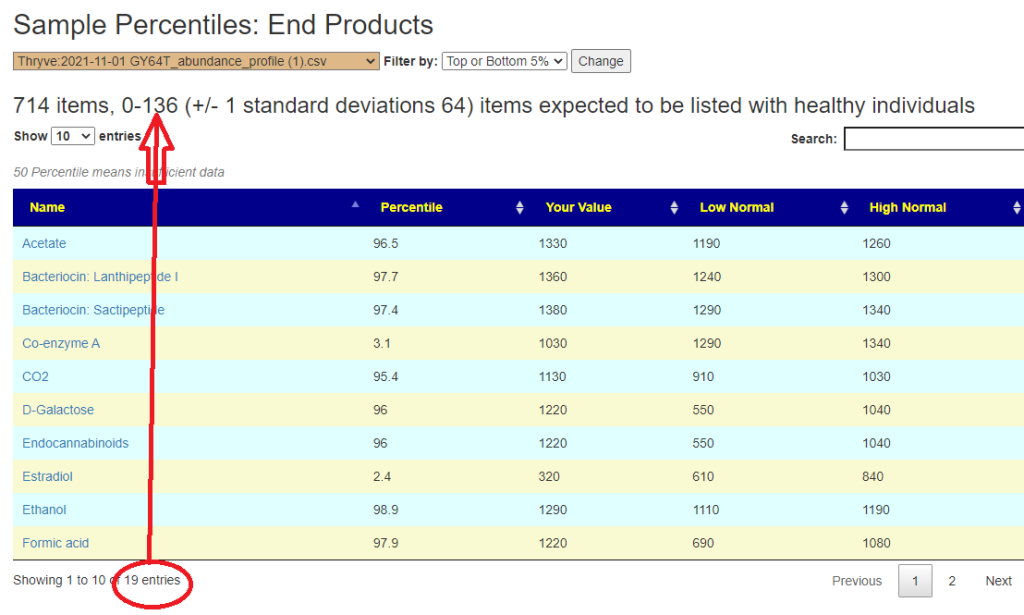
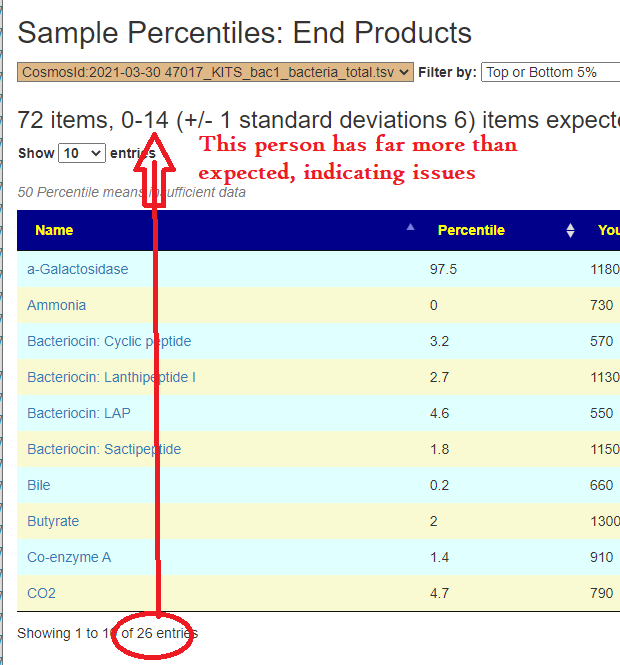
In the earlier version, outliers and full data was on two separate pages. These has been combined into a single page with more options.
Are your abnormal?
Calculations are done for you to indicate the level where values may be out of range by random chance. This is illustrated below.
Display Level: Public — there are no options, top and bottom 5% is predefinedExample of clear evidence of microbiome issues. It looks like the production of Bacteriocins (natural antibiotics) is low, as well as low bile and butyrate production.Display Level: Beginner – The Kaltoft-Moltrup ranges are an available filterDisplay Level: Intermediate – Custom Percentiles ranges are an available filterDisplay Level: Advance- All values is an available filter (this can also be done by setting Custom Percentile to 50%)
Bottom Line
I am hoping to have testing completed by the end of March and will then deploy
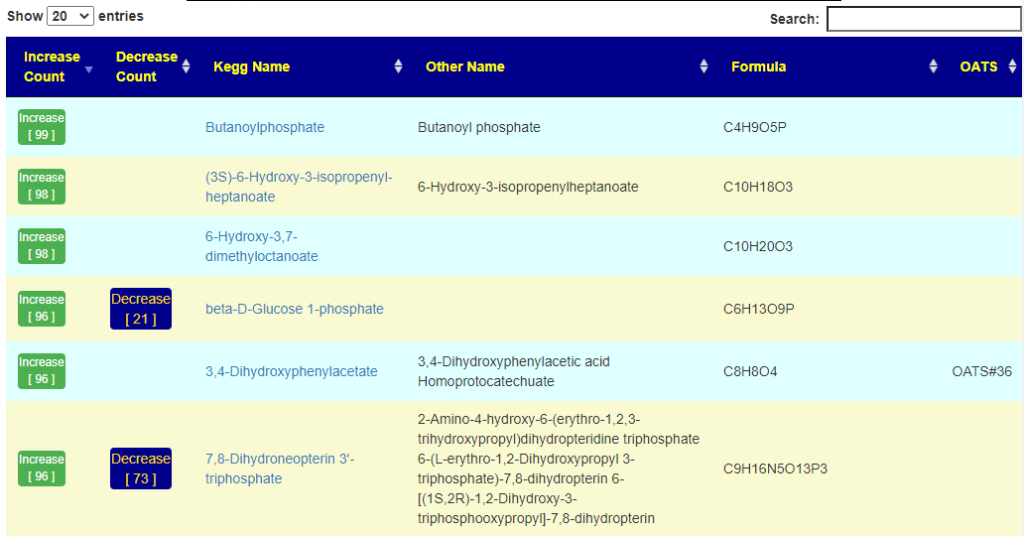
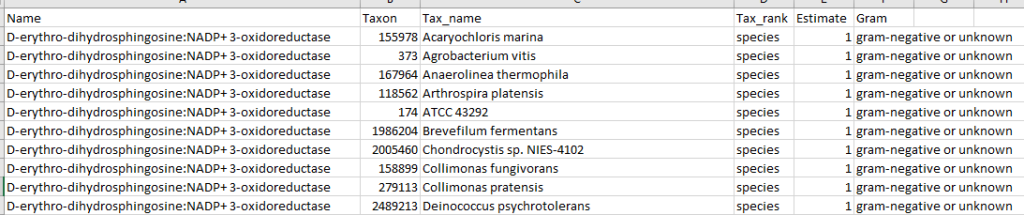
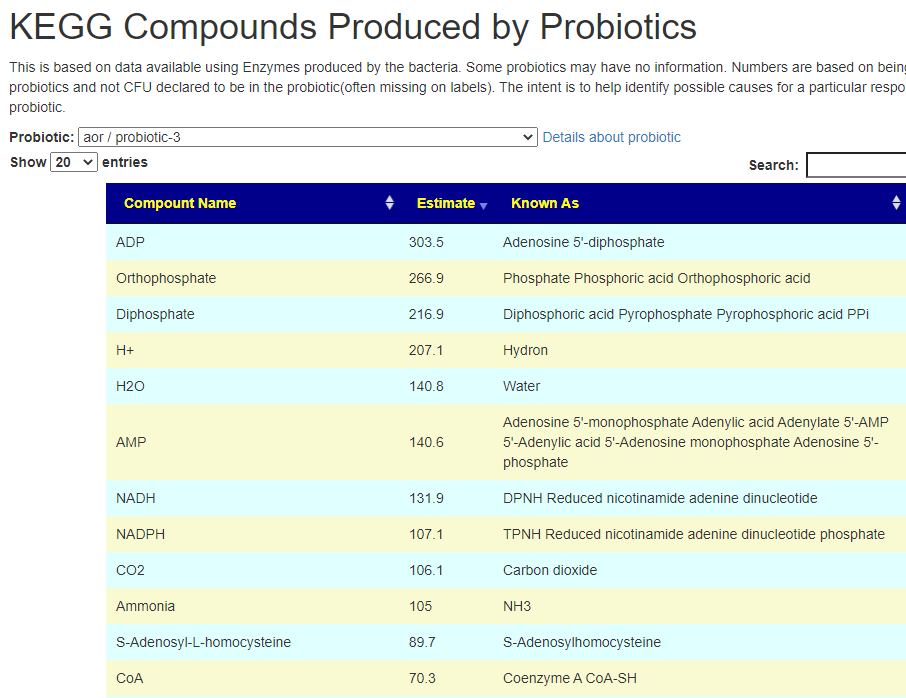
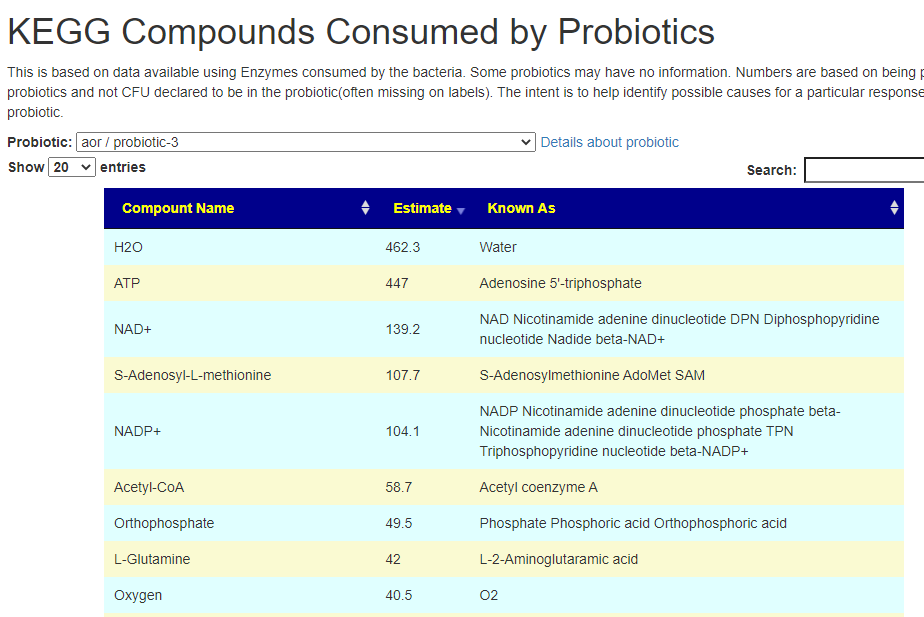
Fortunately due to the data on KEGG: Kyoto Encyclopedia of Genes and Genomes we know what products that most species produces, and can infer the quantity of these products/enzymes that are produced.
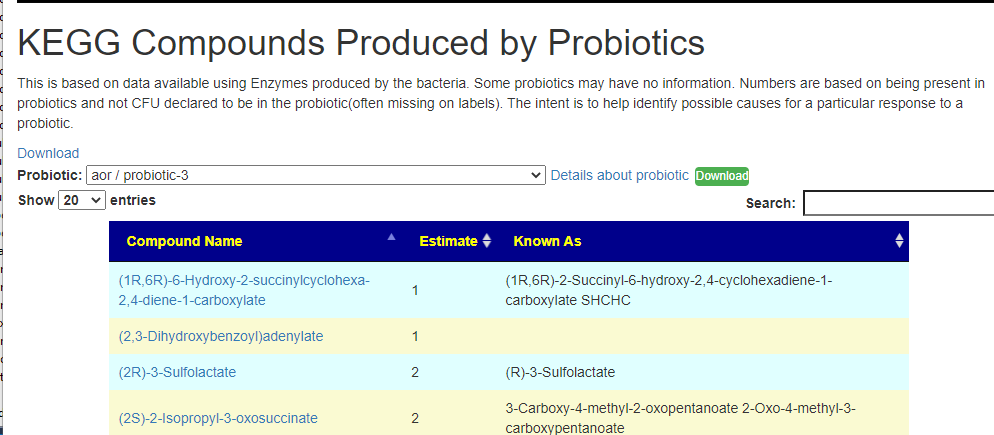
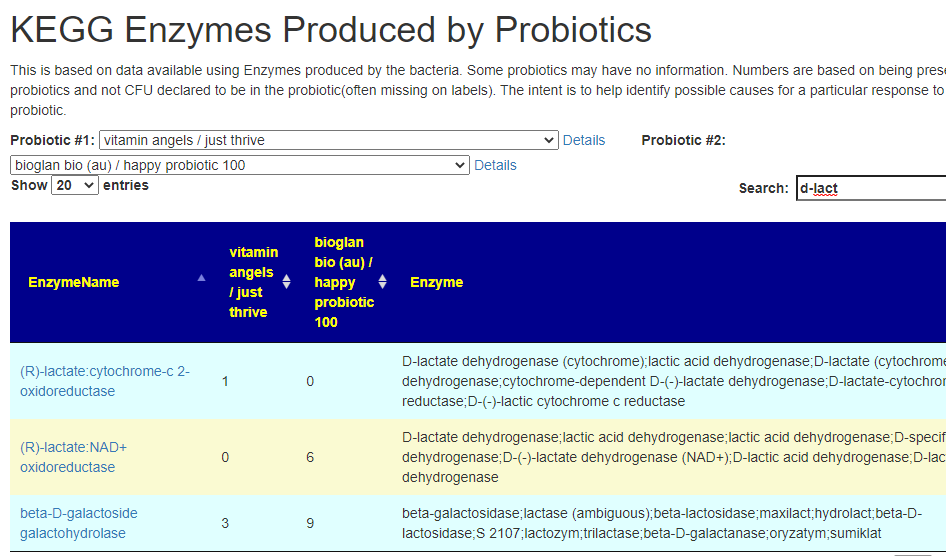
After login, change display Level (top left corner) to a higher level, you will see a new menu item appear with several new pages. This post focuses on the probiotic only.
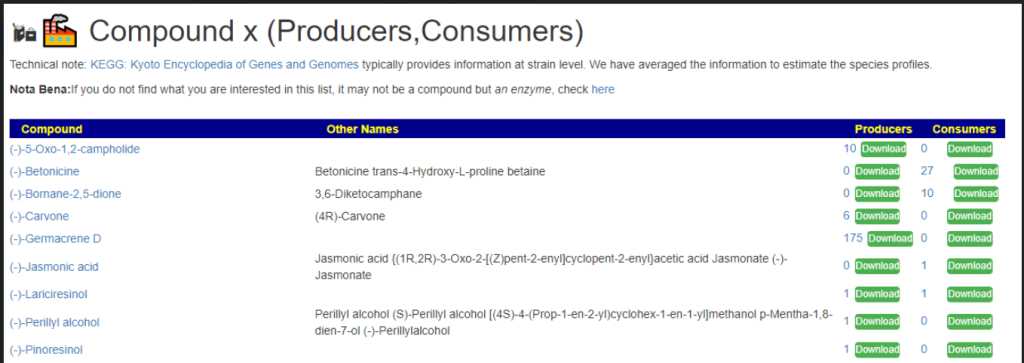
Each page follows the same pattern. Compound listed on left, estimate of number of units produces by cell and alternative names. The Compound or Enzyme is link to the KEGG page providing more (usually very technical) information.
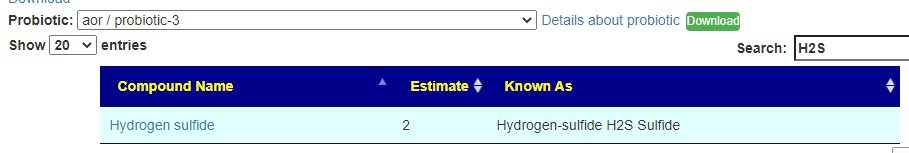
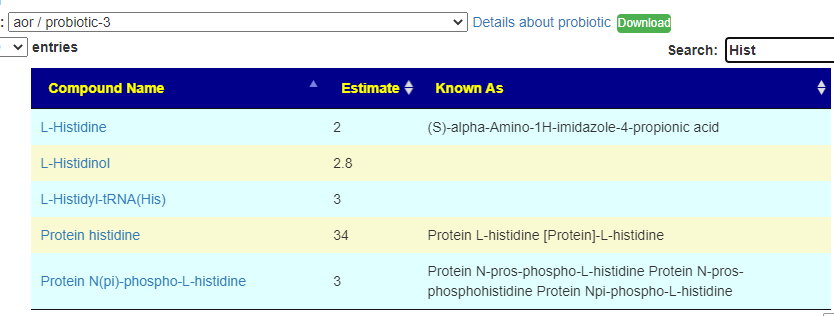
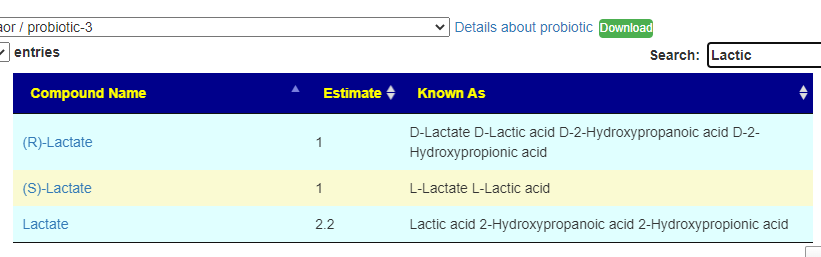
You can search by just typing the name in the search box. Some examples:
Does Probiotic-3 produce Hydrogen Sulfide?Does Probiotic-3 generate histidine (which is then converted to histamine)Does Probiotic-3 produce D-Lactic Acid (which is associated with brain fog)
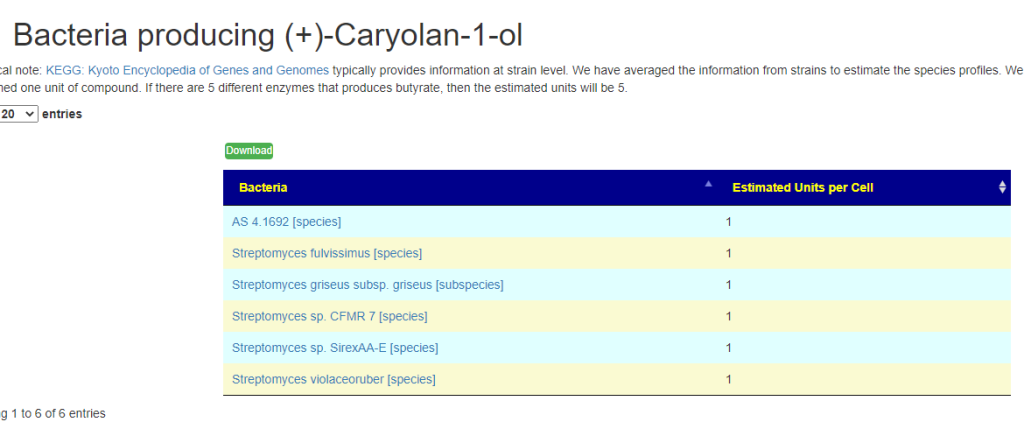
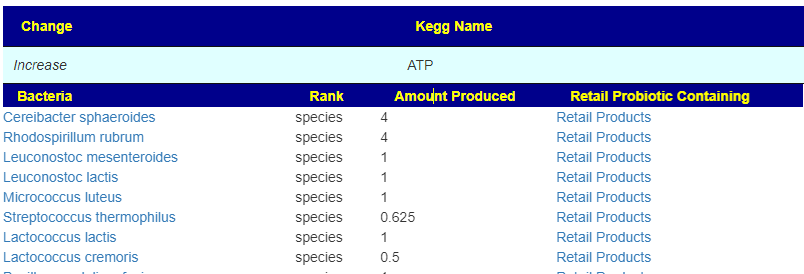
Fast identification of Probiotics for specific purpose
This page shows the compound with links to lists of probiotics. On the far right is the ID from the Organic Acts Tests (OATS – where a match could be identified)
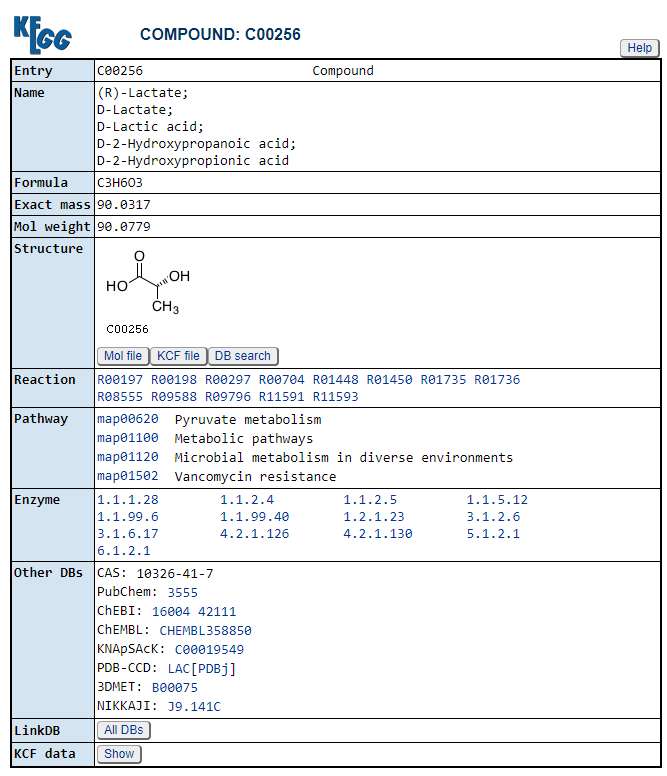
An example of the linked to KEGG page is shown below. This page can be a good starting point for a long and steep learning curve.
Compare Probiotics Pages
These pages allows you to select two different probiotics and see what each produces.
Bottom Line
This provides information that bridges the gaps in published research. If you know what you are trying to increase or decrease, this should provide guidance for you to discuss with your medical professional.
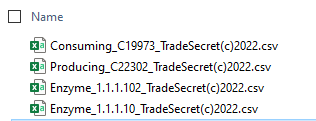
Tonight I implemented some downloads of data as Comma-Delimited files which may typically just be double clicked and open in Excel. This information is intended for researchers who wish to develop and test their own models from the data available. If you have to ask what these are, you are a non-nerd. See KEGG for support, not me.
Note that the files download have “Trade Secrets” and (C) 2002 in the name indicating that further distribution of this data is not permitted without written consent. By accessing the data you consent to those restrictions and acknowledge the nature of the information received.
As some of you are aware, I have spent the last few months refactoring how I handled data from KEGG: Kyoto Encyclopedia of Genes and Genomes. This weekend, I just finished pushing the data (over 8 millions rows of data) to the web site.
The computation is based on for each cell Sum(Sum(enzymes x compound produced by enzymes) over species), and the same number of cells of each species.
Nota Bene: There is a lot of rewiring from old tables to new tables that is in process. Some site pages may break.
A reader asked for my opinion on this. This is one of those topics where emotions often run hot and reason can be forgotten. I usually avoid these topics — I hate flame wars.
All of these passion topics, tend to have the following critical factors:
If those that advocate also sell the product, there is a clear conflict of interest. I will usually assume that their primary motivation is profit that is clothed in the appearance of wanting to help.
If they do not sell, but often use it, then we have the nasty issue of rose color glasses, selected data picking of their patients. They may have a vested interest in wanting to be right or a pioneer. They usually ignore those that have adverse results (often those people do not return to the people treating — hence they do not see these adverse results or discount them to non-compliance to the treatment plan (blame the patient syndrome).
Last thing is desperation, especially when standard of medical care fails — people are willing to try anything, regardless of the risk and often very low success (which could have happen at random).
These risks applies to most items where feelings run high.
First item, is that there is risk: “Soil-transmitted helminth infections represent a large burden with over a quarter of the world’s population at risk. Low cure rates are observed with standard of care (albendazole)” [2022]. Hence, viable (living) helminthiases should be avoid. Killed ones (for their chemicals) would be preferred. If things go bad, you are looking at low cure rate!
Helminth infection is included in my microbiome modifiers with 130+ bacteria impacted, see this page (I also include Round-Up! To include it in your suggestions, pick “Prescription – Other“. Remember NONE of the modifier suggestions should be done without medical review).
Please note also that if the treatment including killing them afterwards, that treatment alone may be responsible for positive effects
“There was evidence of treatment-specific effects among the selected studies, such as findings of treatment-associated taxa, including Sphingobacteriaceae and Flavobacteriaceae (26); increased and decreased Actinobacteria and Bacteroidetes, respectively, after placebo comparison (18); mebendazole treatment-induced changes in the diversity and abundance of Collinsella and Blautia (27);”
The reader referred me to Lindsey Wells, a naturopath, page on Helminth Therapy. The Hygiene Hypothesis of Autoimmunity is cited which I have touched upon in the past 2015, 2018. To me, many advocates of this hypothesis both over simplify and commercialize it. If you wish to take this hypothesis seriously, then there is only one treatments: live on an organic farm, with a lot of different animals — if your boots are not covered in manure, every day — you are not taking the hypothesis seriously!
“The Amish and Hutterites are U.S. agricultural populations whose lifestyles are remarkably similar in many respects but whose farming practices, in particular, are distinct; the former follow traditional farming practices whereas the latter use industrialized farming practices….Despite the similar genetic ancestries and lifestyles of Amish and Hutterite children, the prevalence of asthma and allergic sensitization was 4 and 6 times as low in the Amish” – i.e. industrialized farming practices resulted in six times (600%) the rate of asthma and allergies. See Innate Immunity and Asthma Risk in Amish and Hutterite Farm Children(2016). This is also echoed in their farm products!!! Amish and Hutterite Environmental Farm Products Have Opposite Effects on Experimental Models of Asthma [2016]. Given a choice of buying groceries from a Hutterite farm or a Amish farm, buy the Amish (non industrialized) groceries!!!!
The bottom line of this page is simple — not a single clinical study was cited. The link to https://biomerestoration.com/hdc/ and where I would expect studies (under “The Science”), there was not one.
And the other site cited: https://helminthictherapywiki.org/wiki/index.php/Helminthic_therapy_research had links to a lot of self-published papers (i.e. not peer reviewed) and just 2 on PubMed.
As I stated at the start, this is among the worst form of study because it is prone to the placebo effect plus discontinuation of patients that are non-responders or who have adverse results. They do mention “1% of paediatric patients experienced severe gastrointestinal pains”. No objective measures (labs, etc) were cited. IMHO, a purely subjective report. Note that the journal that it was published in was Journal of Helminthology (I wonder if there is a bias with those doing peer review?)
“Thus, it should come as no surprise that eradicating helminths can result in expression of diseases influenced by these pathways. There are now many animal models representing a diverse range of diseases for which helminths either prevent and/or reverse ongoing pathology. “
“I hypothesized that a treatment with Trichuris suis soluble products represents a feasible holistic treatment for autism, and the key for the development of novel treatments. Preclinical studies are required to test this hypothesis.”
Bottom line: there are no clinical studies supporting it use for autism, it is all theoretical. Microbiome Prescription does include it in the options of gut modifiers — so you can objectively see if it is a good fit for an autistic child’s microbiome.
A Critical Criteria: If something has been proposed for 5+ years and fail to produce a positive clinical study then assume there have been several studies done with no positive results.
To me, it is not a rational choice — significant risk of known demonstrated adverse issues with no significant demonstrated positive impact. Odds are that going camping for 2 months in the woods will have a greater positive impact, or better still, work on an organic farm tending chickens, pigs and shoveling manure! Getting involved with 4H may be a good thing in many ways!
Warning: This is for Advance Users and Microbiome Nerds This year I have been updating KEGG data with an careful audit. The existing KEGG data wasstrain specific, and the numbers being generated were only for those strains. I have averaged out over the species that these strains belong to so I can compute KEGG data compensating for missing data. Most 16s labs are reasonable complete for species reports but report only a few strains.
The result will be more accurate estimates when I update all of the pages and data computed from the raw data upload.
I am a firm believer in openness — showing which study or source that I obtain the data from. This allows people to independently validate (or nit-pick applicability).
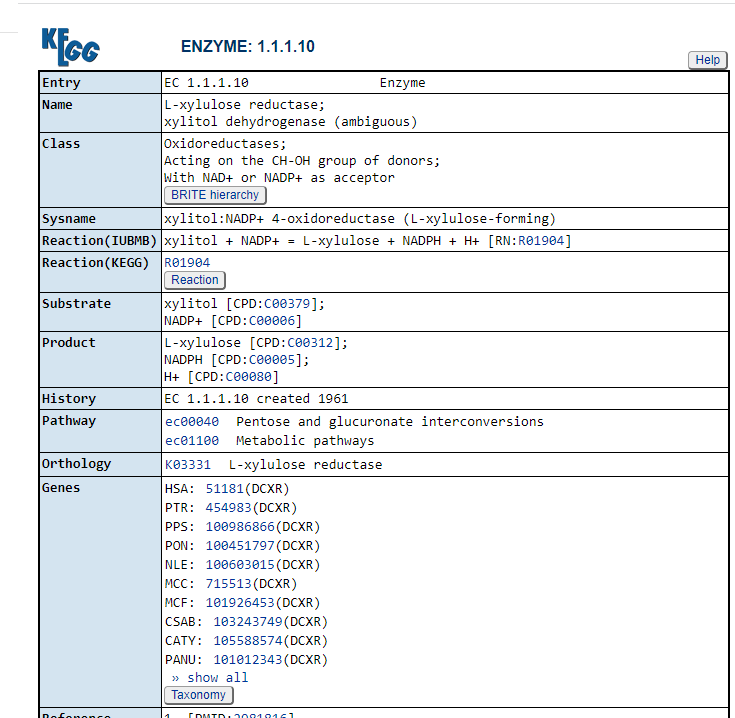
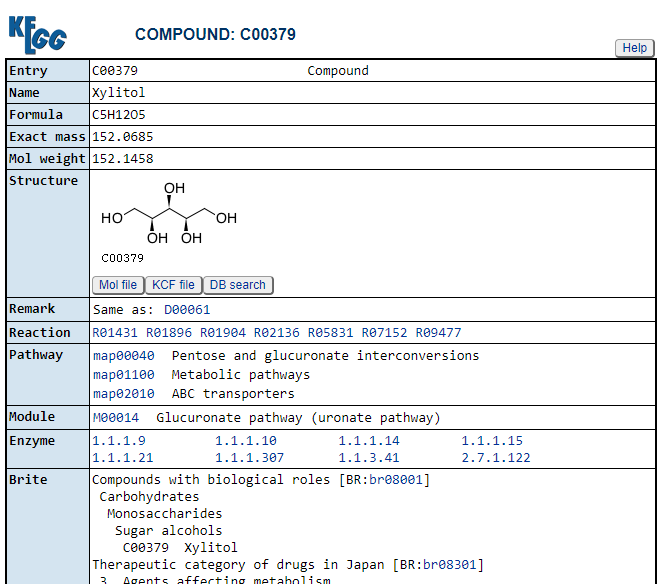
Clicking on the compounds (CPD: ….) takes you to a description of the compound
https://www.genome.jp/entry/C00379
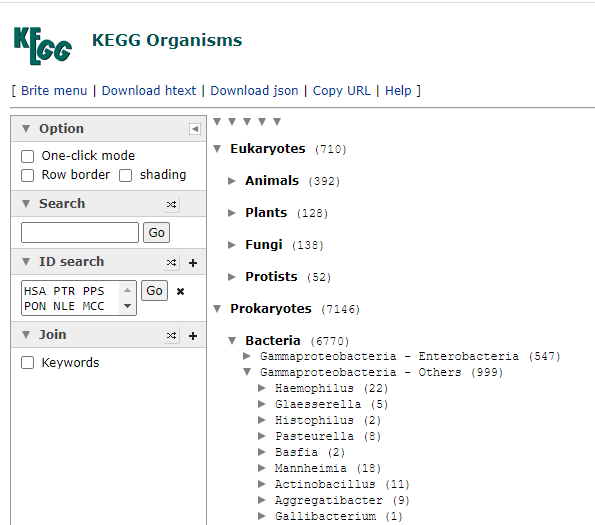
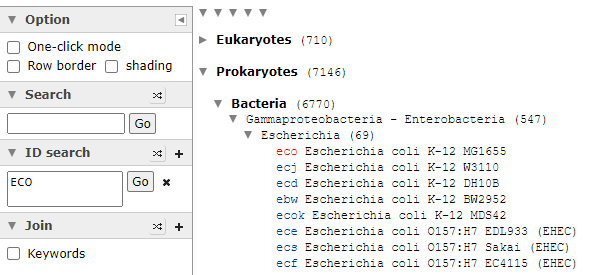
Then click on Taxonomy to get the bacteria (and other things it is found in). This will show you an expanding and collapsing tree. Look for Prokaryotes / Bacteria
Expand until you find a strain of interest, all strains will be listed, it is the ones that are in blue, items in red do not have this enzyme. As you see below, this approach has some technical issues, one strain of Escherichia Coli has an enzyme and most of the others do not. To be technically complete, we would need the labs to identify every strain of Escherichia Coli as well as KEGG having all strains — that is unlikely for decades…
Clicking on it will show you the bacteria details as shown below with the all important NCBI Taxon number.
What is happening under the cover…
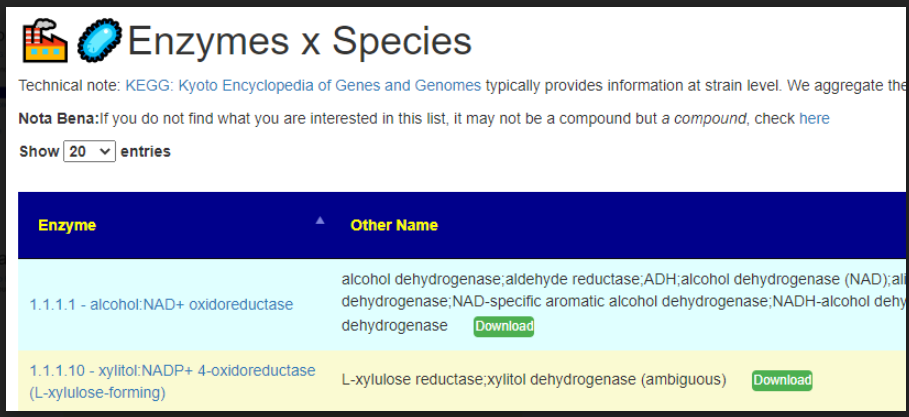
All of this data is extracted and re-aggerated into 3 main pages:
In the case of drill downs to bacteria, we also show a count of the number of enzymes in the bacteria that produces or consumes a compound, as illustrated below:
Step 2 is applying it to the samples and re-computing the totals and percentiles. Needless to say, many of these pages will be hidden on the menu to the casual user because it would result in information overload. An advance display level will be required. The above links should always work.
I still have some tuning to do, there are a few thing slightly off… but what is there is reasonably accurate.
A reader with ME/CFS (thus with brain fog) was unable to figure out how to get antibiotics and other (off label) prescription drugs.
This person has a willing MD and in consultation with him, they are wishing to try the Ceclie Jadin protocol that has had over a 70% success rate (when properly done) with ME/CFS. I said “properly done” because I have seen the antibiotics being used continuously (instead of one week on, three weeks off) and without rotation by some MDs that think they can do better (with no clinical experience to support it).







Recent Comments