For a time, the answer to this question was straightforward: uBiome.com was the go-to choice for microbiome testing. However, uBiome went out of business in 2019 following legal and regulatory issues, including an FBI investigation and bankruptcy1 3 . As a result, uBiome is no longer an option.
Today, for most individuals—especially those not working directly with a specialist—the best available test is offered by BiomeSight.com (There is a discount code “MICRO” that may still be available). Unlike many other platforms, BiomeSight allows users full access to their raw data, supports data uploads from other providers, and offers advanced tools for those who want to explore their results in depth. Critically, they have the easiest transfer of data to Microbiome Prescription.
The reasons for this recommendation are detailed below.
Declaration of Interests
I do not sell any products or offer any services, nor do I have any financial interest in any microbiome testing companies.
My experience with microbiome dysbiosis, both personally and within my family, motivated me to develop Microbiome Prescription. Initially, I wrote code for my own use on my personal computer. Over time, I transitioned the platform to the web so that it could be accessible to everyone.
I offer data on Microbiome Prescription freely to individuals out of compassion for the suffering of others and as part of religious obligation-duty. Feel not! I will not preach.
An extreme shortage of expertise
The typical medical doctor or naturopath receives surprisingly little training on how to influence or manipulate the microbiome—often less than what you could learn from a focused day of watching YouTube videos. Most of their education is based on memorizing standard treatment protocols, which they then apply to patients in a “cookbook” fashion. For example:
- If a patient has Crohn’s disease, prescribe these drugs.
- If someone struggles with insomnia, suggest these herbs.
The core issue is that treatments are usually prescribed based on the diagnosis alone, rather than being tailored to the individual’s unique microbiome. In effect, practitioners are relying on broad stereotypes rather than personalized care.
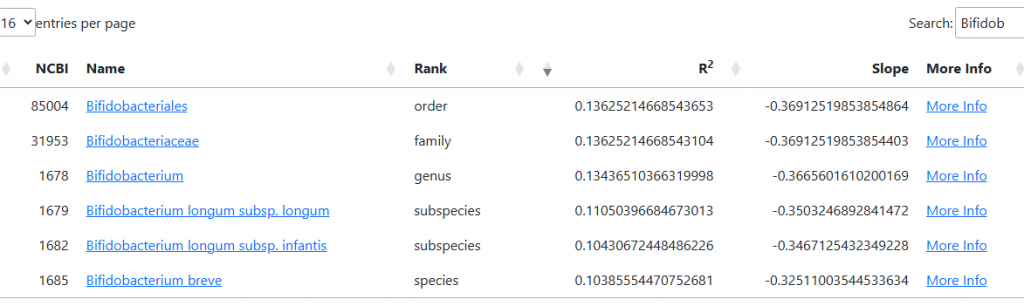
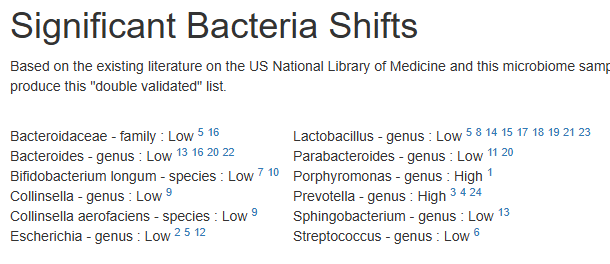
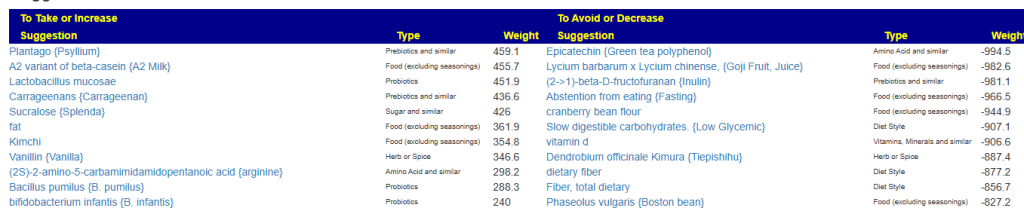
Many healthcare practitioners have limited familiarity with the vast diversity of gut bacteria, often focusing narrowly on well-known genera like Lactobacillus and Bifidobacterium due to gaps in training and the complexity of microbiome science. Practitioners will often focus on these because they know no better. They do not know the significance of high Sphingobacterium bambusae or Dolichospermum curvum.
First Question: Who will interpret the results?
There are common paths that people take:
- The lab themselves.
- If they sell any product, there is a huge conflict of interest between their financial health and your personal health!
- Often their suggestions are based off trolling the internet or using existing AI search engines
- Most AI is prone to hallucinations
- Often they may be based on a single study found on the US National Library of Medicine
- I know one startup in this area that has a team of 6 Ph.D.s (or candidates) building a superior database than what Microbiome Prescription gas. They are barely 10% done. That is a very significant cost and needs significant financial backing to do. It requires a strongcommitment to not do a “good enough” to keep investors happy.
- Word of warning: uBiome.com was one of the earliest labs and dominated the market. They filed for bankruptcy, IMHO due to investor pressure to turn a profit. Many labs keep expenses to the minimum and do not do due-diligence.
- The healthcare practitioners that you are using and a specific test that they recommend. This assumes that they have been trained on the test. Buyer Beware: many will keep to cook book recipes.
- You will go off and find a consultant to interpret the test. Conceptually that sound wise. The problem is that there are so many tests out there and finding someone with experience and successful track record is likely very hard.
- You take ownership and education of the issue using selective resources, for example Microbiome Prescription free site. You should review your plans with your regular medical professional to insure no clear adverse issues with you medical history. In short, you are not asking them to develop a plan — you are asking them to make sure this plan is reasonable with no known health risks.
Amount of information in Test Result
The table below gives the possible test types. Typically:
- qPCR gives 30-100 bacteria
- 16S gives 600 bacteria
- Shotgun gives 6000 bacteria
IMHO 16S is the best give what we know about how to change the microbiome.
| Method | What It Measures | Strengths | Limitations |
|---|---|---|---|
| 16S rRNA Sequencing | Bacterial taxonomy (genus level) | Cost-effective, widely used | Limited resolution, misses non-bacteria |
| Shotgun Metagenomics | All DNA (all microbes, functions) | High resolution, functional insights | Expensive, complex analysis |
| qPCR | Specific DNA targets | Rapid, quantitative, targeted | Limited scope, requires known targets |
| Culture-Based | Viable, culturable microbes | Functional studies, viability assessment | Misses unculturable microbes |
| Metatranscriptomics | Active gene expression (RNA) | Reveals microbial activity | Technically demanding, expensive |
| Metaproteomics | Proteins produced | Functional protein insights | Complex, needs advanced equipment |
| Metabolomics | Metabolites (small molecules) | Functional metabolic readout | Source of metabolites often ambiguous |
| Specialized Sampling | Microbes from specific locations | Spatial resolution, targeted sampling | Invasive, technically challenging |
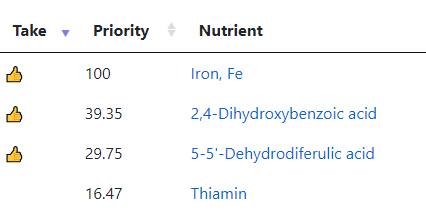
What test gives the most actionable items using Microbiome Prescription database and expert system?
As a statistician, sample size determines the ability to detect patterns, make forecasts and thus identify the key bacteria connected to your symptoms.
- BiomeSight has 4382 samples
- OmbreLabs/Thryve has 1530 sample
- Every other labs has less samples.
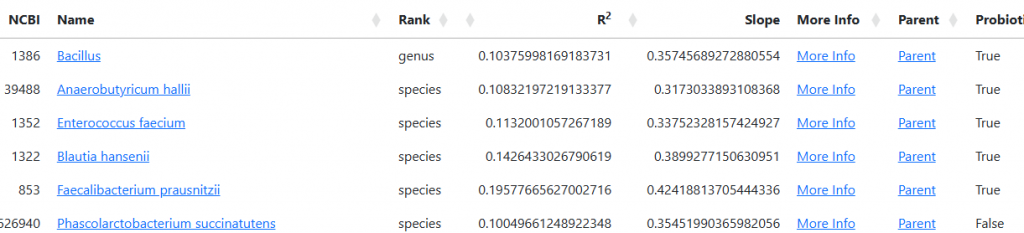
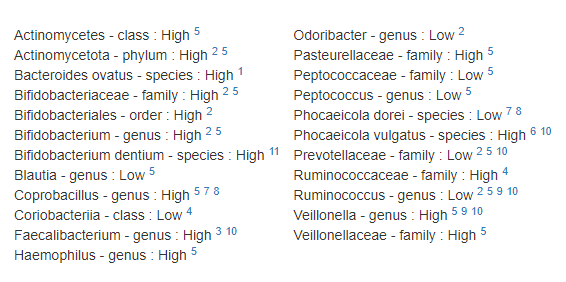
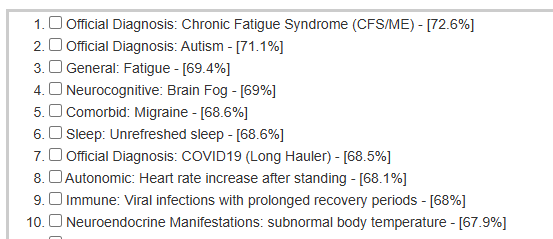
See Bacteria by Symptoms by Lab that are high or low for symptoms that can be accurately predicted from a microbiome:
- BiomeSight has 308 Symptoms – thus we know which bacteria of the 600 to focus on
- OmbreLabs/Thryve has 86 Symptoms
- uBiome has 67 symptoms
There are a few things that 16s does not detect which shotgun does (virus, phages, antibiotic resistance etc). A small number of people may need that information … unfortunately, the ability to detect which bacteria are responsible for your symptoms is lost.
For more background, you may want to watch this video.
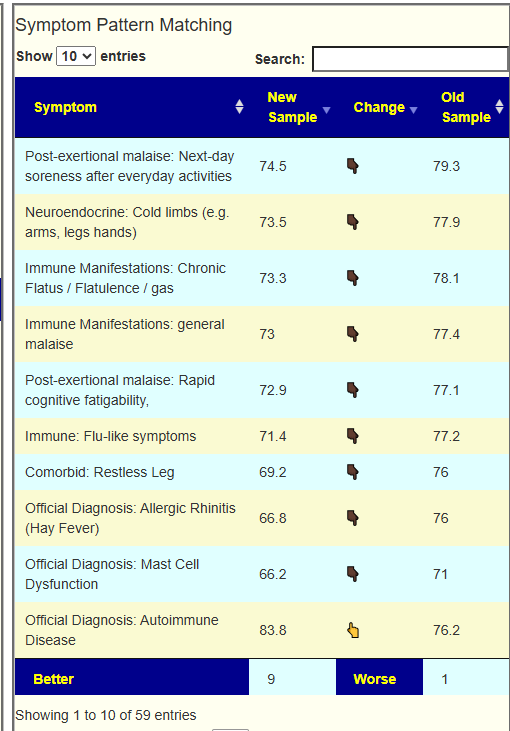
This recent discussion with a Long COVID patient that used Biomesight and the data from Microbiome Prescription may be helpful.












Recent Comments