For my last of the 4 installment per weekend, I will look at computing statistics. I have found non-parametrics analysis to get more interesting results, but classic core statistics are also helpful to understand the nature of each taxonomic layer.
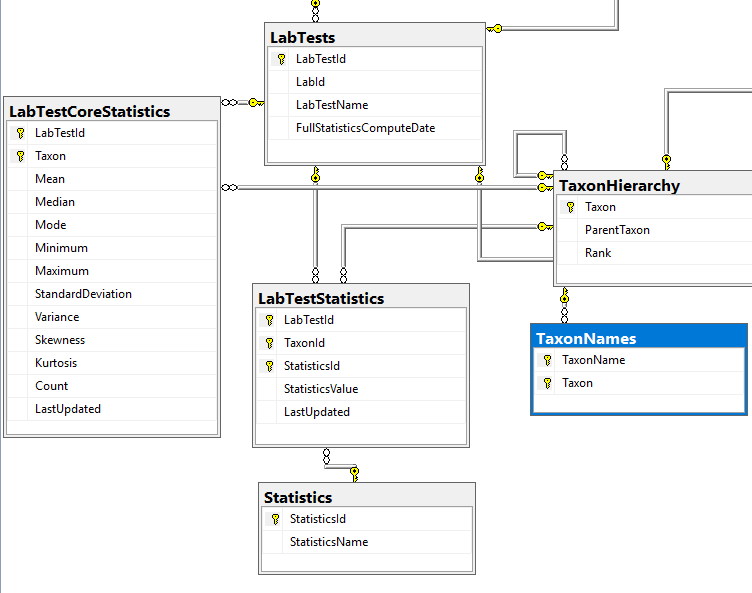
Data Schema Update
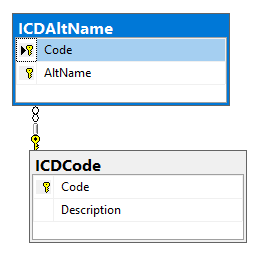
We are dealing with this section of the schema which has another table added.
For thoses who are not statisticians, terms like Kurtosis may sound like a medical condition and not a statistical measure. I have enclosed links to Wikipedia articles on each of these items below.
That’s it for this weeks installments. Homework on this one is whether you should include Zero (none-found) values or not. This is actually a complex question which may depend on what percentage of samples have a specific taxonomic unit.
The prior posts dealt with uploading text files – perhaps a csv file or a json file. Sometimes reports are sent as PDF. Today I received a good example of a PDF to show how to upload it.
We use the Taxon Name and Rank process as done in this earlier post. The trick is pretty simple: decompose the file using itext7 or earlier versions. And then create an appropriate data table (same format as in the earlier post), and use the same store procedure.
As before, not found bacteria are written to a file for later analysis of exception. In this case there was only two items not found :-).
Part of being open sources is being open-source on where you are getting information. For myself, my primary criteria is published peer-reviewed scientific papers. You must determine your own criteria (for example, using unpublished studies referenced and summarized in patent filings).
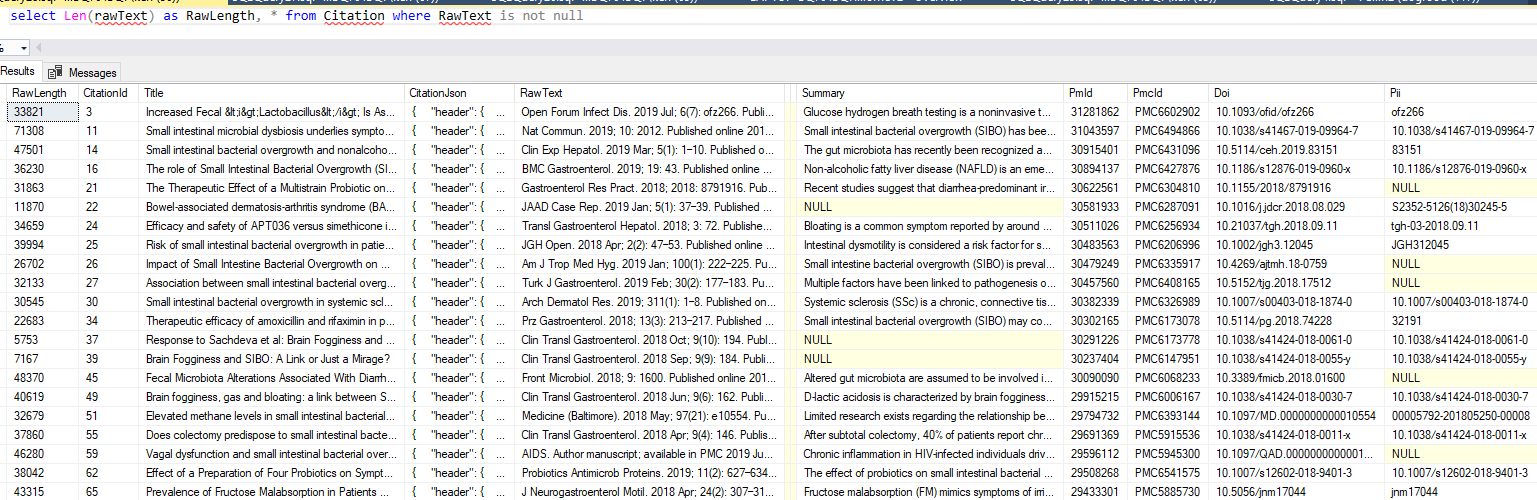
Citations are the connectors between bacteria, probiotics, medical conditions, lab measurements etc. They are the evidence chain of how things are related.
In this post I will describe column by column what I feel should be in your citation table as a minimum:
CitationId – unique identifier used in relationships : int
Title – The title of the study or document: nvarchar(255)
CitationJson – A summary using the PubMed pattern: nvarchar(max)
RawText – A pure text version of the document. It will be used for Text Mining and manual inspection : nvarchar(max)
RawDocument – A binary version of the document. Often this will be a PDF file or may be an image. varbinary(max)
RawDocumentMimeType – The mime type for the RawDocument: varchar(60)
Summary – This is typically the PubMed summary for the study: nvarchar(max)
Ideally, a component should extract the RawDocument into RawText. For example, OCR for images and various converters for PDF. This type of conversion is outside of the scope of this blog series. They can be done (I have written such professionally).
Running it is simple, just give it the name of the condition (it defaults to 100 citations, but you can increase it. I usually run with 10,000 citations at a time for each bacteria taxonomy name).
CitationConsole “Some Unusual Condition”
What is the net result? Running it with “SIBO”, we see in the database.
So we have our data ready for either text mining or manual review.
Home Work
We can now fill up our citation tables, in some cases with the complete text (excluding appendices) as well as a short summary. How are you going to process the data?
We want to view relationships between microbiome and various medical conditions. There is also a need to know the impact of various probiotics on various conditions.
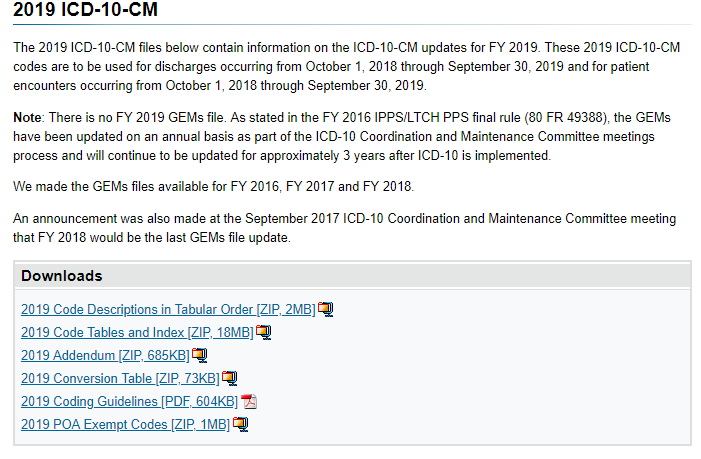
The International Classification of Disease (ICD) is the equivalent as taxon numbers for bacteria and is a well defined comprehensive list of medical conditions. We may need to add alternative names for some of the conditions to match common usage.
If something is not there (it should be), we can always add our own codes and diagnosis. For example there is no “SIBO” or ” Small intestine bacterial overgrowth” item, scanning on like we see that the code of A04.9 is often used. We would insert that into the ICDAltName table (likely two rows, one for each variation).
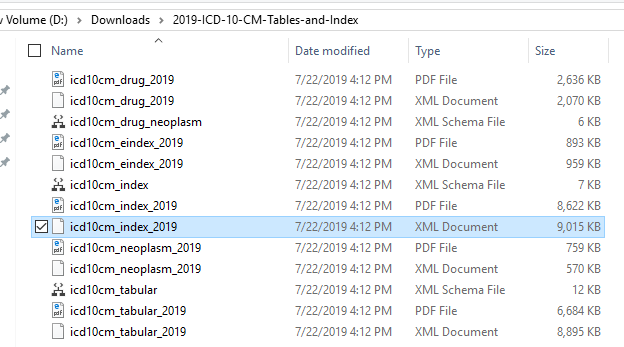
First we need to download some files as shown below.
This file is the parameter to pass to the C# console application that will upload and populate the table. Most of the magic happens in SQL.
We end up with tables as shown below. In the next posts we look at connecting conditions to probiotics to studies, and conditions to microbiomes to studies.
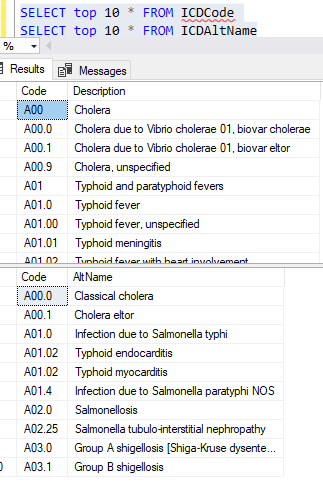
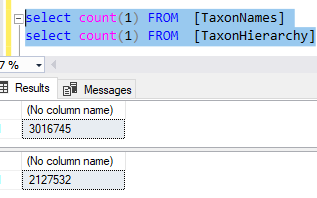
Example of the data after uploading is shown below. There are around 44 thousand IDC codes – fortunately, they are in a hierarchy.
We have some 44 thousand items which are in a hierarchy if you use the code. There are many ways to handle that on a web site — what approach will you do?
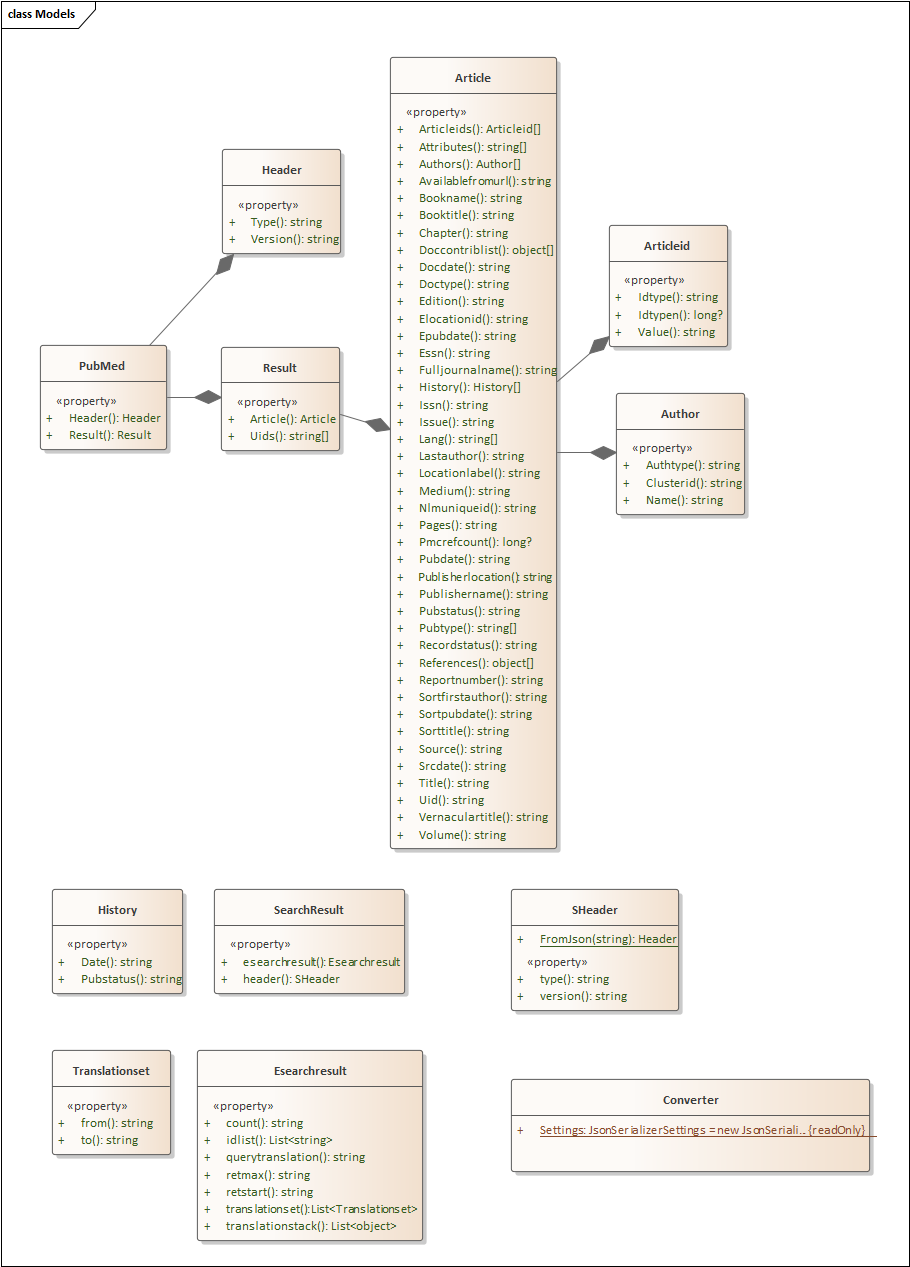
The data format is automatically detected. Classes in the code converted to Interfaces for eventual consolidation into a DLL containing all of the uploads.
The solution was to extract the information into chunks.
A better solution is to get the actual reported values entered. At this point, we encounter the problems of different methodologies at different labs. Technically, the count of one lab cannot be reliably compare to the count at a different lab. There are some ways to mitigate this issue which is outside of the scope of this set of posts (coding).
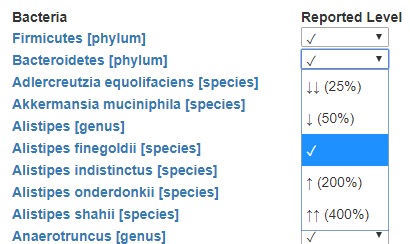
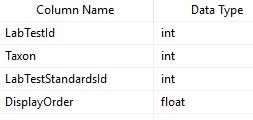
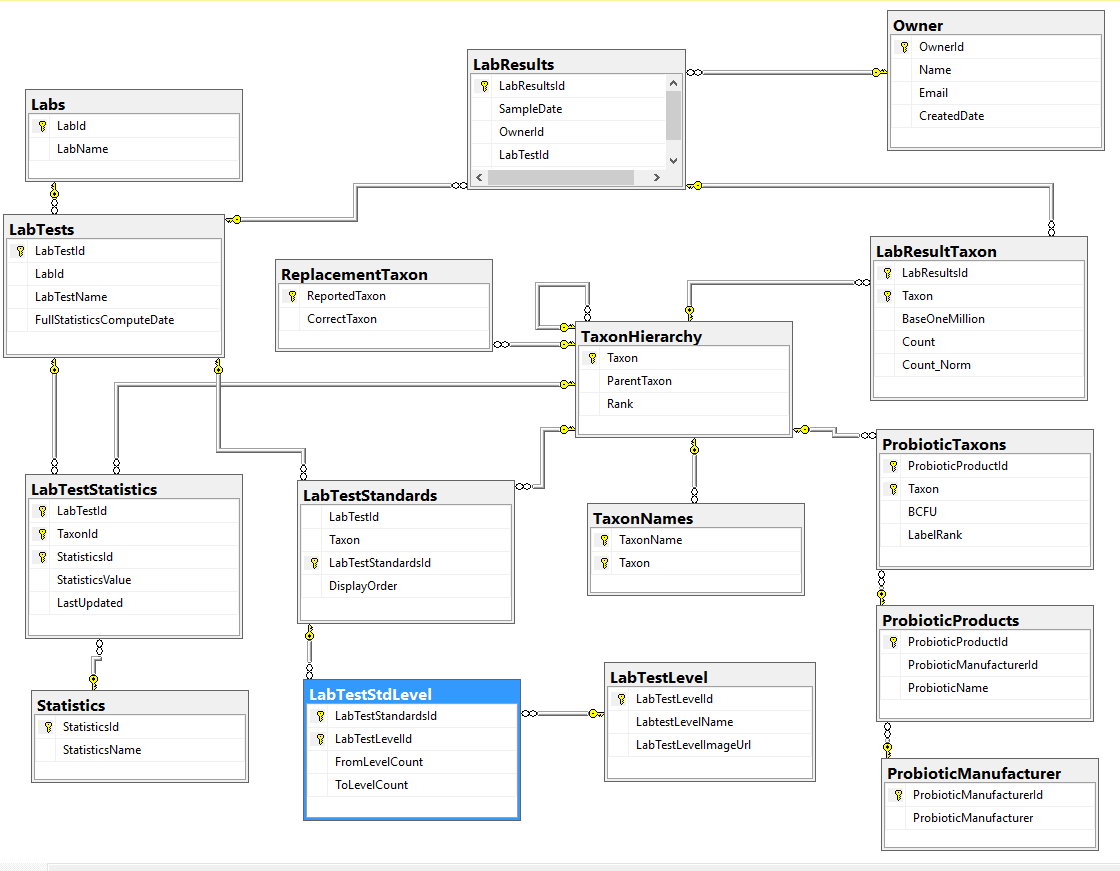
For each of these labs we need to know what bacteria is reported — this would be used done by this new table. We add DisplayOrder so the bacteria will appear the same as on the printed or pdf report (makes entry easier!)
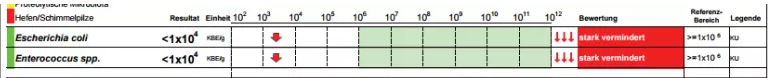
The rest results may be in any of the following presentations
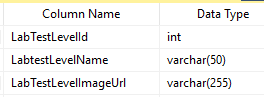
To handle this, we add another table with verbal descriptions and links to images (optional)
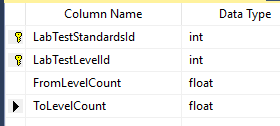
Last thing is to enter the value for each. For simplicity, I opted to define ranges of value.
So, to recap this last table defines the range for a specific bacteria on a specific test that would be deem to be have some label. The data needs to be entered for each bacteria in each test.
Our current database schema
In today’s code example, I enclose a download from my site of the lab tests with their taxons (which may be incomplete or incorrect). I did this by putting into a text file the taxons,displayorder, and then naming the file {labName}.{LabTestName}.txt. They will all be dropped into a /data/ folder.
This makes it easy for lab tests to be revised/updated (and hopefully, with a pull request for the updated date — this is open source, some payback to the project is expected behavior).
After the upload you should see
Select * from [dbo].[LabTestStandards] SELECT * from LabTests SELECT * from labs
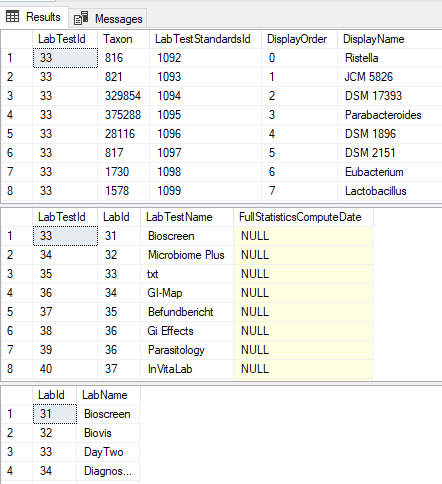
When the Levels and Ranges are added, things becomes a little more complex for processing. If you execute the following and save it as an .xml file (in the /data/ folder), I will write a demo program when I get a pull request on the data. Do it one by one for each LabTestName (GI-Map is shown below) –> GI-Map.xml
SELECT * from LabTests T (NOLOCK)
JOIN LabTestStandards S (NOLOCK)
ON T.LabTestId=S.LabTestId
LEFT Join LabTestStdLevel L (NOLOCK)
ON L.LabTestStandardsId = S.LabTestStandardsId
LEFT JOIN LabTestLevel M (NOLOCK)
ON L.LabTestLevelId=M.LabTestLevelId
WHERE LabTestName='GI-Map'
FOR XML AUTO
The result is XML to be dropped into a file
.... etc ...
There is actually a stored procedure that does it for you: ExportLabSettingsForSharing @LabName varchar(50)=’GI-Map’
There is also one that generates the data to use for data entry:
DisplayNewEntryForm @LabName varchar(50)=’GI-Map’
Bottom Line
This post definitely has a home work assignment — obtain the level data and names from existing reports that you have available (or can borrow from friends).
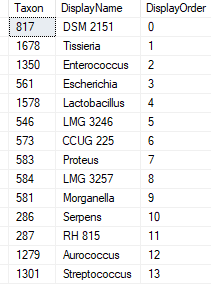
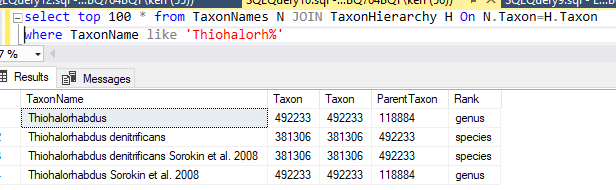
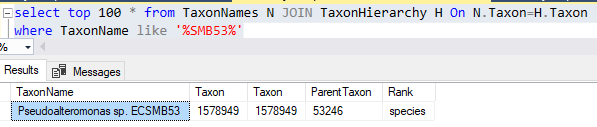
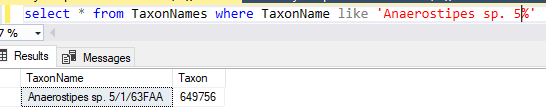
This data table is then uploaded and Sql goes thru a series of steps to attempt to match it. Things that are not matched are written to a file (like done in the prior post). In this case, the solution is much easier to patch the differences. Just add the name to the TaxonNames file with it’s appropriate taxon (no need to create a separate substitution table). The Sql attempts to match by taxon rank, name first, if that fails, it falls back to name alone.
For the test file that I used, there was a lot of mismatches
Spot checking for a few, I discovered that these are currently listed as having a parent of “unclassified Gammaproteobacteria” (118884)
For others, there is nothing that seems to be a match and the original data line does not clarify anything.
For a 103 line input we got 87 matches and the rest unclear. The sample code gives you the information on the challenges. You need to come up with your own resolutions (discard or patch)
P.S. Remember to update your SQL Server Schema to get the new sprocs and data types.
I have just added another console application. I am doing things in layers and downstream we could create a DLL with everything in it. At this point (especially for those wishing to port it to other languages), doing one feature in one console application is likely the best approach.
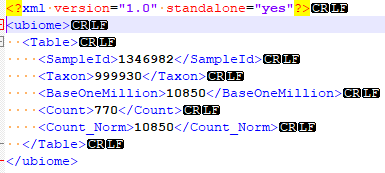
This installment takes the ubiome json file and uploads it to the tables.
This is the template to use for uploading tests that provides the standardized taxon number. The only different would be in file parsing.
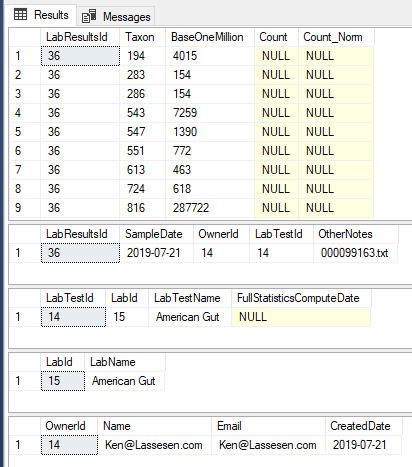
After one upload, your data should look like this:
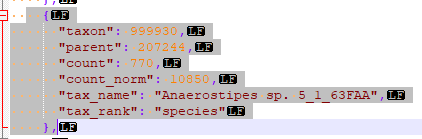
The code is simple, a single method that reads the file name, takes the JSON and makes an object. Walks the object and create a data table. Then calls a stored procedure with this data table and other information.
One thing that is also done is it writes a report on any taxon that it could not match to the ncbi microbiome hierarchy. “Different strokes for different folks”. Actually, more often a taxonomy got deprecated and ubiome has not updated their system.
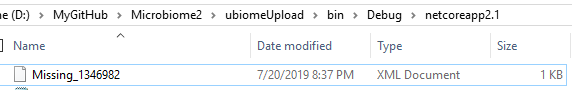
In the execution folder, you will see a file containing the missing taxon
With the contents like:
If you open the json file, and search for it, you will see what they call it.
You could add this to the taxonHierarchy or ignore it. I searched for it by name and got an apparent match but with a different taxon number.
Resolving this disagreement is up to you. One option is a replacement table of ubiome’s taxon to ncbi taxon where you are confident that they are the same.
Readers have expressed interest in some of my work being open sourced. The actual site would be described as an “evolved beta”, rather than subject people to quirks and kludges, I am proceeding as a redesign of a V.2 product. If you are interested, please FOLLOW (top left) to get updates as they happen.
The Repository is at:
https://github.com/Lassesen/Microbiome2
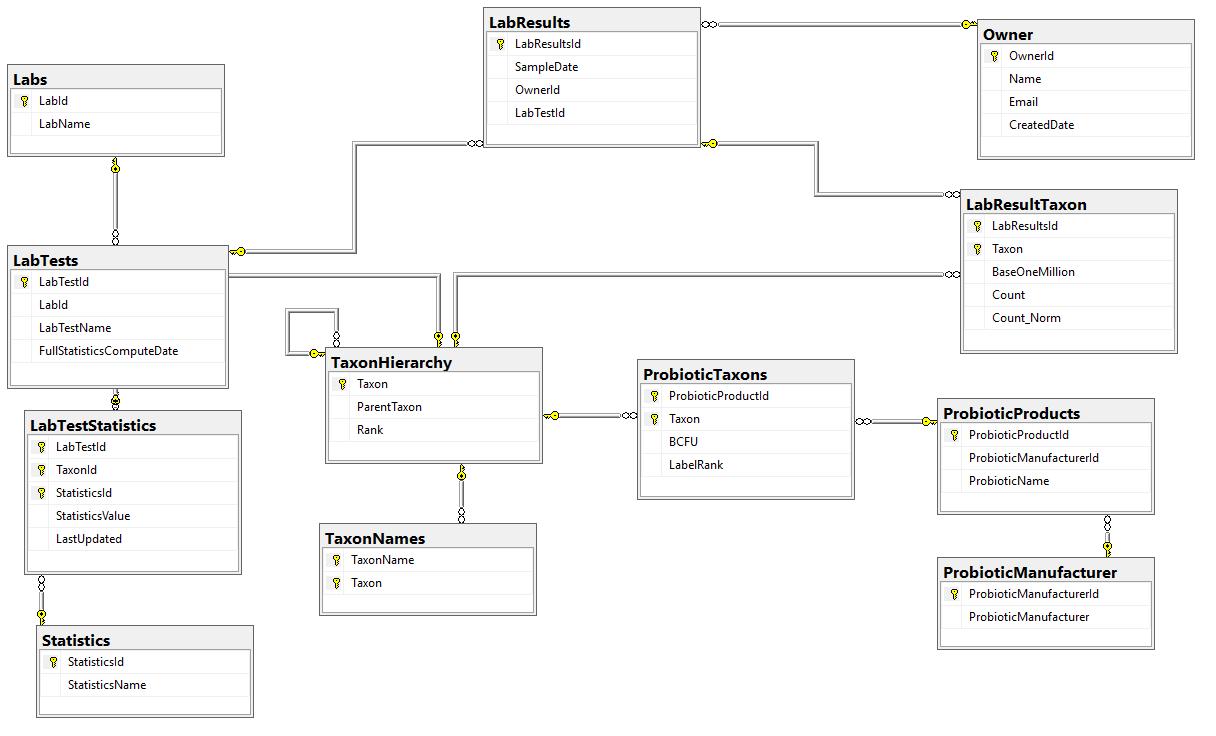
The first item that I want to get up for discussion is the core database tables – for review and comments. The Database diagram is shown below.
A few quick notes:
Statistics were done as a separate table instead of the typical additional columns because trying multiple quantiles is seen as the way to go for non-parametric analysis. This becomes open ended with items like “Q2_18” – Quantile 2 of a 18 way quantization being possible. With that type of breakdown, we want to know if we are dealing with stale date, so we need to know the computation date.
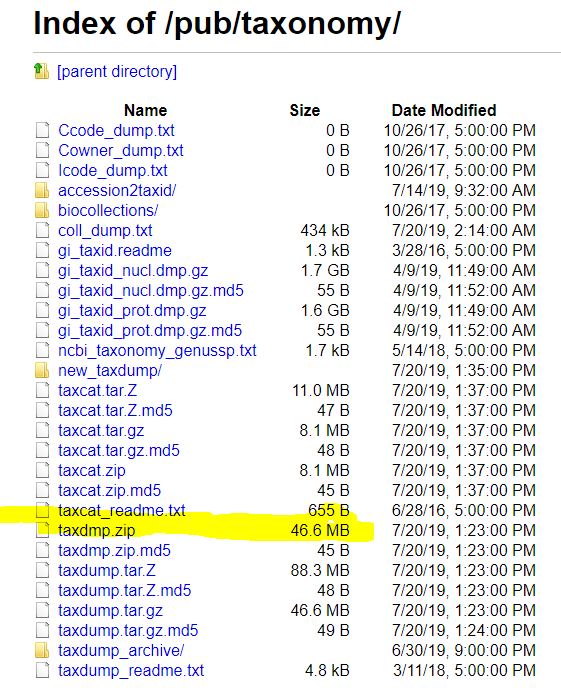
Next post will deal with populating TaxonHierarchy and TaxonNames from ncbi downloads.



Recent Comments