See also:
Several readers have emailed this article (or news story on it) Current microbiome analyses may falsely detect species that are not actually present. or The virtual microbiome: A computational framework to evaluate microbiome analyses. This is not a surprise, it was reported earlier
Common approaches to analyzing DNA from a community of microbes, called a microbiome, can yield erroneous results, in large part due to the incomplete databases used to identify microbial DNA sequences.
The process is equivalent to naming a person’s last name from a random DNA sample of a person.
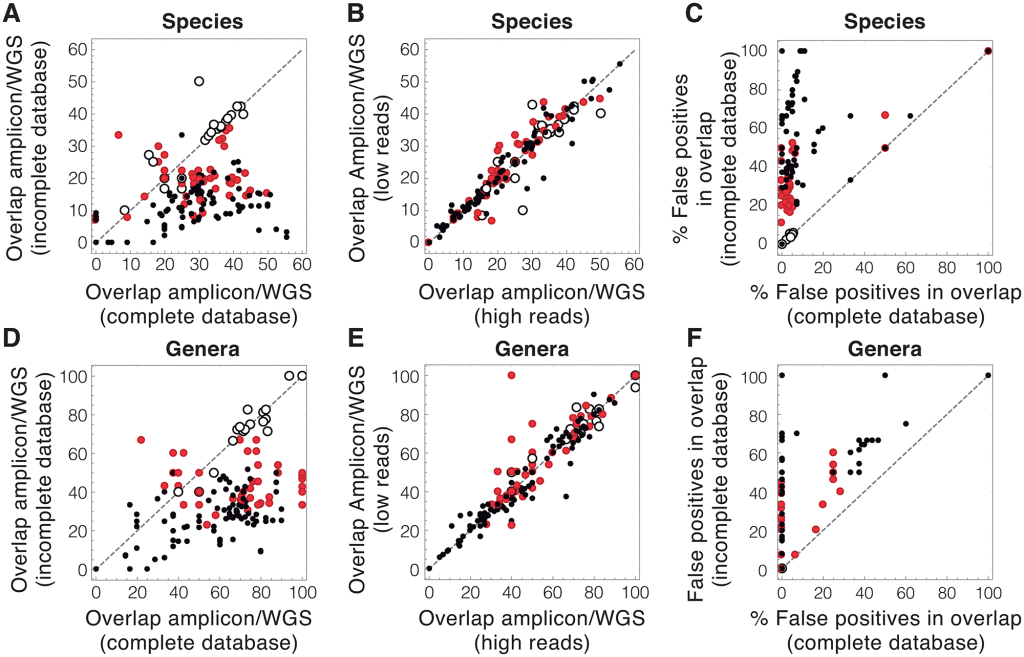
To reduce the uncertainty of microbiome data, the effort in the field must be channeled towards significantly increasing the amount of available genomic information and optimizing the use of this information.
The analogy of “The process is equivalent to naming a person’s last name from a random DNA sample of a person” is a good description of the issue. If you get more people DNA is the database, the odds of correctly identifying the person’s birth last name increases…. naming the bacteria species or strain has the same issue.
For the purposes of Microbiome Prescription, it is not a significant factor because the Artificial Intelligence is based on odds and probability (just like finding the name). For a human, you may identify that it is likely a Norwegian or Dane and thus the last name likely ends with a “sen” with 4.6% odds of being a Jensen (see more here). It is significant if your ideology requires absolute answers.
The script of the first Episode from 2019 is repeated below.
The taxonomy nightmare before Christmas…
This post is intended to educate people more on the technical aspects of the microbiome. I am not talking about taking 4 samples from one stool and sending it to 4 different testing company. I am talking about one sample sent to one testing company which then provided their analysis and a FASTQ file. The raw data.
What is a FASTQ file (besides being megabytes big)? It is the DNA (technically the RNA) of the bacteria in the stool. It looks like this (using the 4 letters that DNA has):
CCGGACTACACGGGTTTCTAATCCTGTTTGATACCCACTCTTTCGAGCATCAGTGTCAGTTGCAGTCCAGTGAGCAGCCTTCGCAATCGGAGTTCATCGTTATATCTAAGCATTTCACCGCTACACAACGAATTCCGCACACCTCTA
The file that I am using as text would be around 16 megabytes. This data comes from a lab machine. The company then processes it through their software to match up sequences to bacteria.
In this post, I am using the FASTQ from uBiome and getting reports on the bacteria from:
- ubiome
- thryve inside
- biomesight
- sequentia biotech.
Naively, one would expect almost identical results. What I got is shown in detail below. At a high level we had the following taxa counts reported
- ubiome – 253
- thryve inside – 632
- biomesight – 558
- sequentia biotech 366
I did a more technical post on my other blog. From some providers, a taxonomy may be 40% on another 2% or even none… ugly!

The headaches!
Number One Issue: You cannot, repeat cannot, compare a taxonomy report from one lab with another. EVER!
- I have 8 uBiome reports and 2 Thryve reports. I can compare the uBiome to each other and the Thryve to each other. I can never mix their direct taxonomy reports !
Number Two Issue: If I wish to compare different lab reports, I MUST obtain the FastQ files from each lab and process them thru the same provider. The FastQ files are the raw data! For me, I prefer to push them through multiple providers which means that the 10 reports suddenly become 40 or 50 different reports in my site.
- This means a lot more work for the typical user. It also means that guidance, like that from Jason Hawrelak Criteria for Healthy Gut, would need to be revised to be provider specific!
For more details with examples, see The problem with “official” ranges from labs
My Headaches
I have revised my site to show data by specific provider (while keeping the across all provider data still available). A lot of pages to revise and test.


4 thoughts on “The taxonomy nightmare — Episode II”
Comments are closed.