Over the last few years, I have been trying to tease relationship out of data. I have tried a wide variety of methods and finally found one that been producing good results.
The method is conceptually straight forward:
Take the actual reading and apply a monotonic increasing function to it. Thus if Valuea < valueb then func(Valuea) < func(valueb)
With the resulting data, transform it to be a rectangular distribution for all samples
Hypothesis test the values from people who recorded symptoms using P=0.01 as a threshold
Once the candidate association are done then we can also test if a sample’s item satisfies the hypothesis.
This approach has some nice characteristics, because it will detect patterns that:
are not linear on the values
does not assume a normal distribution
does not not assume items are caused by end associations (i.e. too high or too low)
In some cases, we see a shift into a middle range that is statistically significant
Adjusting “Middle Peak” patterns
Both of the above above are typical beliefs that people will attempt to apply to the data.
Comparing uniform distribution to normal distribution
I have refactored Bacteria Interactions Why? on the site to use the data discovered via this post. The information is more accurate and more comprehensive than the prior version
I have done a quick demo, shown below, and I will add a few examples after.
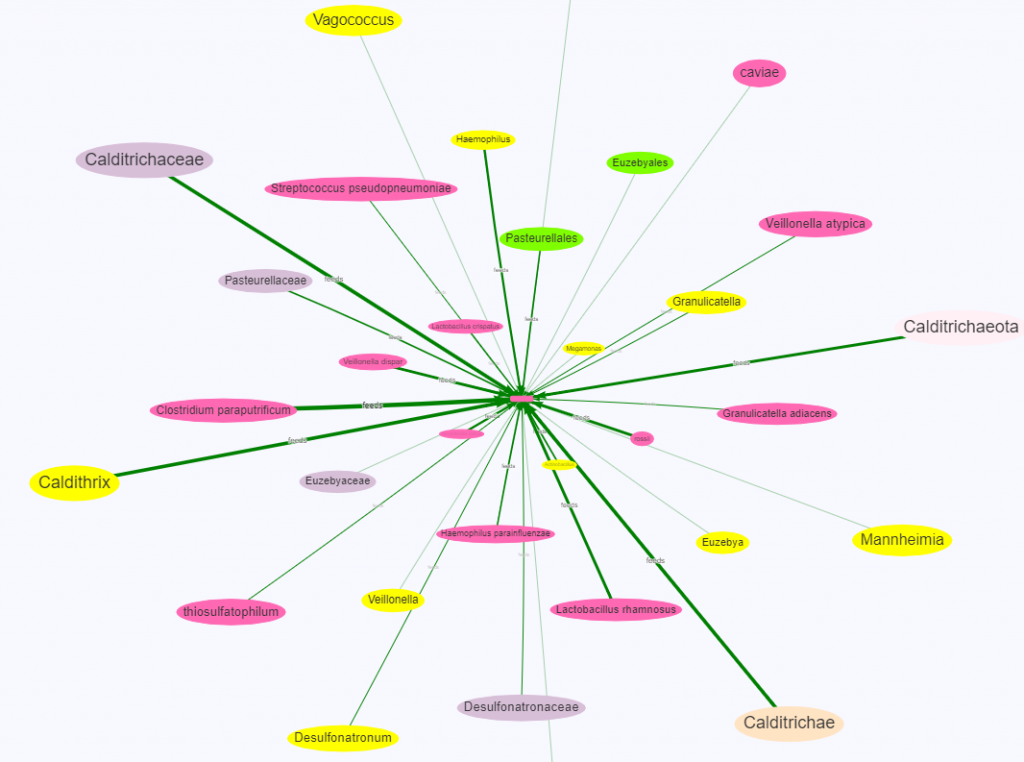
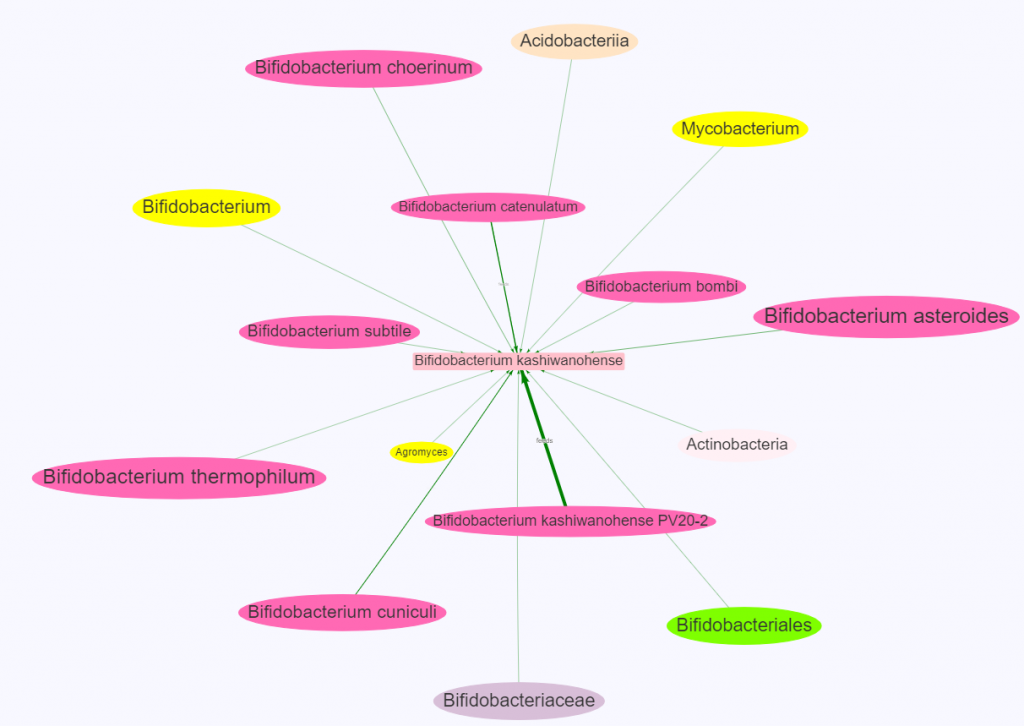
Key points
The color of the oval indicate level of hierarchy
Same color to the middle one indicates that it’s independent
Often other colors are children or parents
Line thickness indicate amount of influence
Size of the ovals indicate percentile ranking.
A small oval indicates less than bacteria than normally seen
A large oval indicates more bacteria than normally seen
Examples
In our first example below we see many other species encourages this species If we look at it’s hierarchy on NCBI, we see a lot of bacteria that are not related by descent.
For the next example we see many siblings influencing it.
Bottom Line
Beyond the fun to see aspect, if there is an item of concern you may wish to see what bacteria influences it and include those in a hand-picked sample.
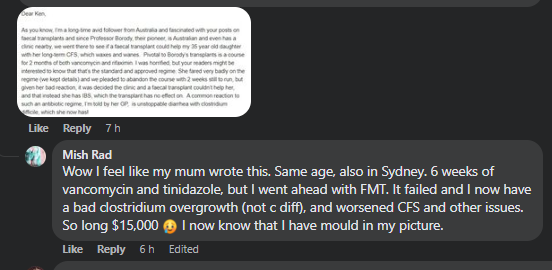
…did a NirvanaBiome test before and 4 weeks after finishing the FMT. As far we can see her gut condition only got worse.”
On The Gut Club: Stool Test Discussion Group Facebook group, some other comments were shared
My first goal is to try to understand what went wrong and how. Then looking at “where do we go from here”. Ending with a reading list on FMT.
Additional Personal Experience with FMT shared
Personal Email (with permission)From Facebook
Analysis of Changes
I started with comparing Bacteria/Taxonomy Out of Range Over Time which showed 63 items. The most interesting are below. Percentile means where in a collection of 2000+ samples that the reading is. For example 97%ile means that 60 samples had more than this and 1940 samples has less – most people would deem that to be an excessive overgrowth. Prior means before the FMT, After means weeks after the FMT.
This dramatic shift may be (1) a defect in the classification algorithm or (2) a bug in my specific import routine for CosmosId [which I am looking at) or (3) sibling families taking over [siblings tend to like the same environments] , I am inclined to the first cause(1) since the data is pushed through the same code(2) but (3) is almost as probable as (1). See my 2019 The taxonomy nightmare before Christmas… post for background.
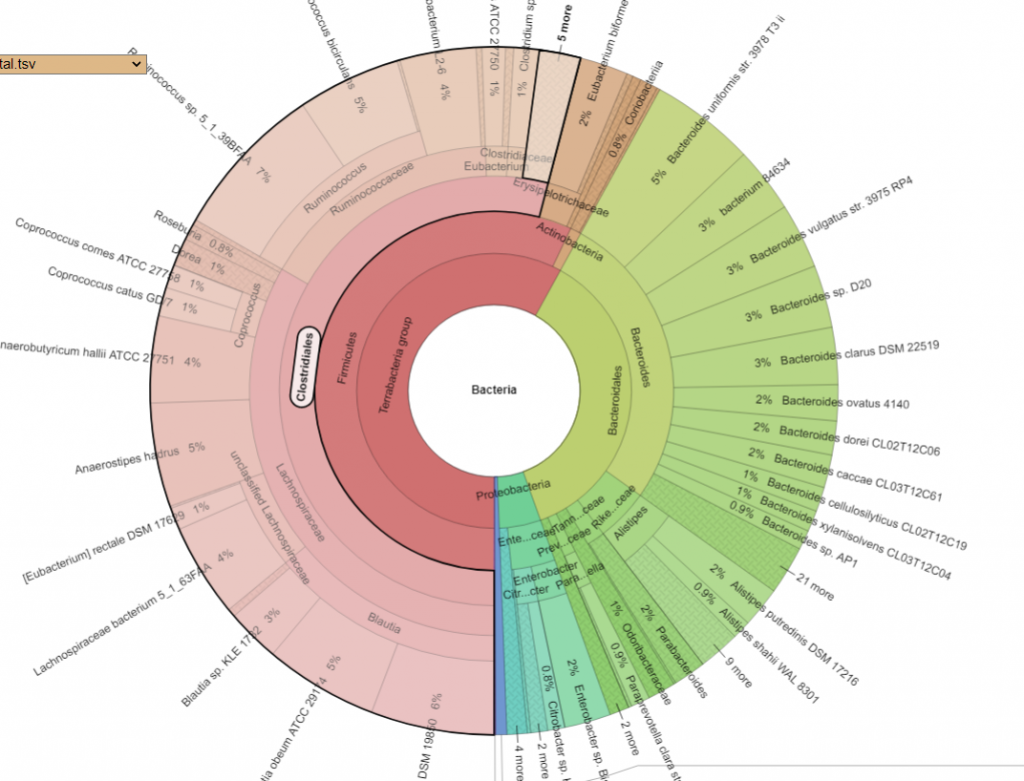
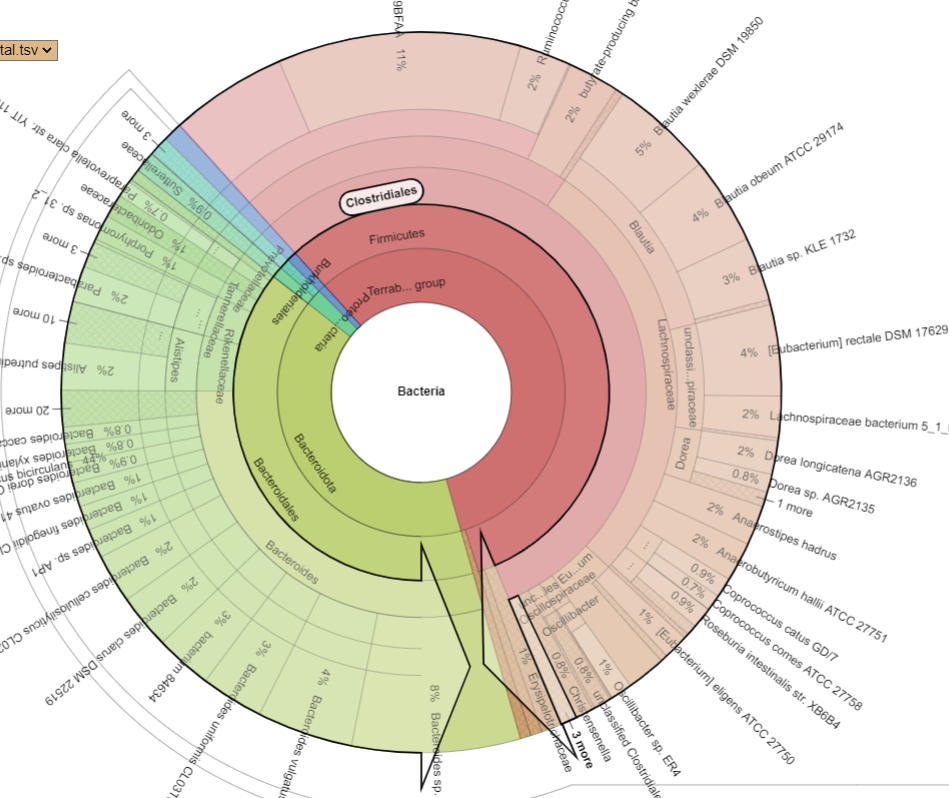
Pie Charts
The two charts below show the growth of Clostridiales and the reduction of every other order. I have often described Fecal Matter Transplants as equivalent to an Organ Transplant (or Blood Transfusions) with the same issues of rejection being significant. We do not know yet now to “type” or test the new item for compatibility.
I speculate that there may have been warfare with several orders weakened, Clostridiales was already near an extreme value — my gut feeling is that bacteria with strong overgrowth are dominated by strains that have the following characteristics
They are more robust (i.e. more resistant to bacteriocins (natural antibiotic) produced by other bacteria
They produce significantly more strong bacteriocins
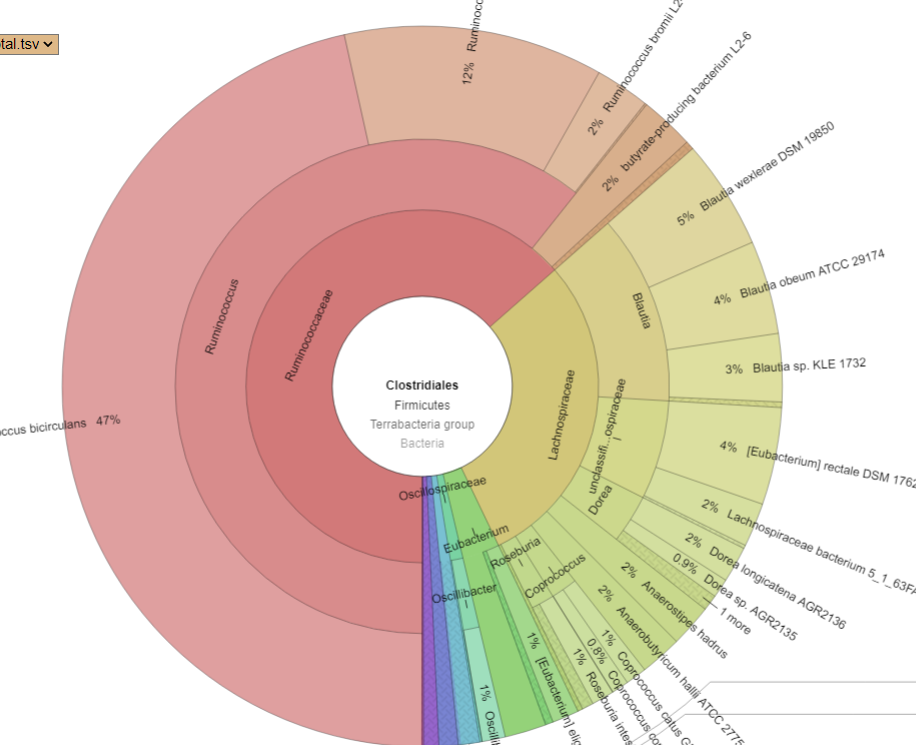
PRIOR: Clostridiales is 54% of BacteriaAFTER: Clostridiales is 94% of Bacteria
The diagram below helps to explain bacteriocins. It show different strains of Lactobacillus Reuteri. Reuterin is the bacteriocins(natural antibiotics) produced. Some produce a high amount, some a low amount and others none.
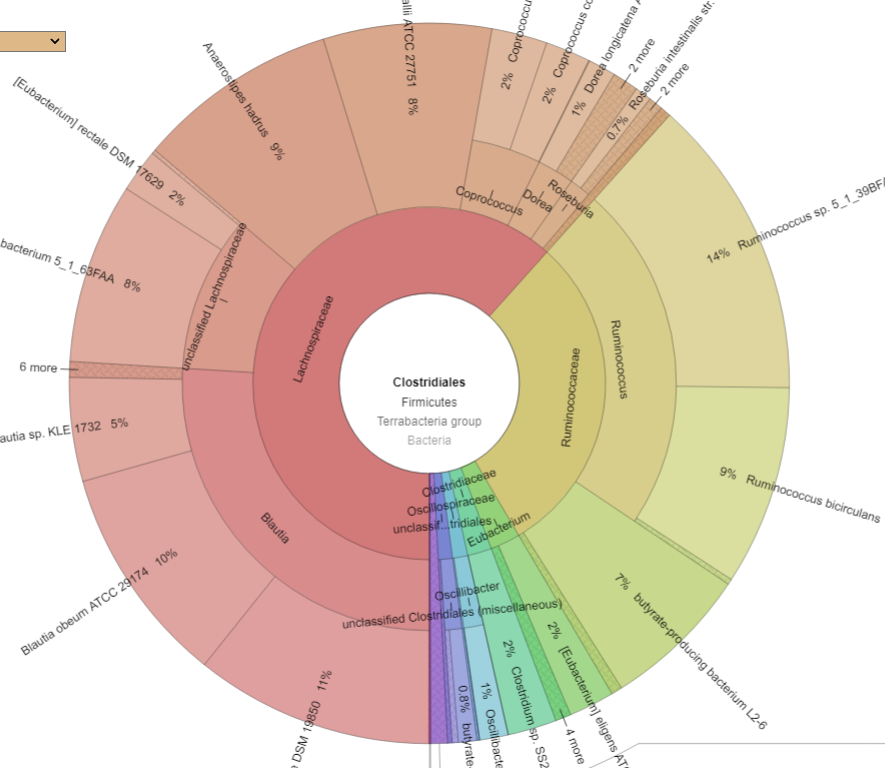
Drilling down into Clostridiales we see major shifts. CosmosId report by strains, so these numbers are reliable
PRIORAFTER
What we see ins that one species EXPLODED, Ruminococcus bicirculans. Prior to the FMT, it’s count was 47,810 or 0.5%, after it jumped to 444,900 (44.5%!!!) – a 10x increase. Whether it was a new strain that was introduced by the FMT that the existing microbiome could not handled as well as the donor (I deem more likely) or an existing strain that filled the vacuum resulting from the FMT bacteria and native bacteria wiping each other out (less likely) is speculation.
Suggestions
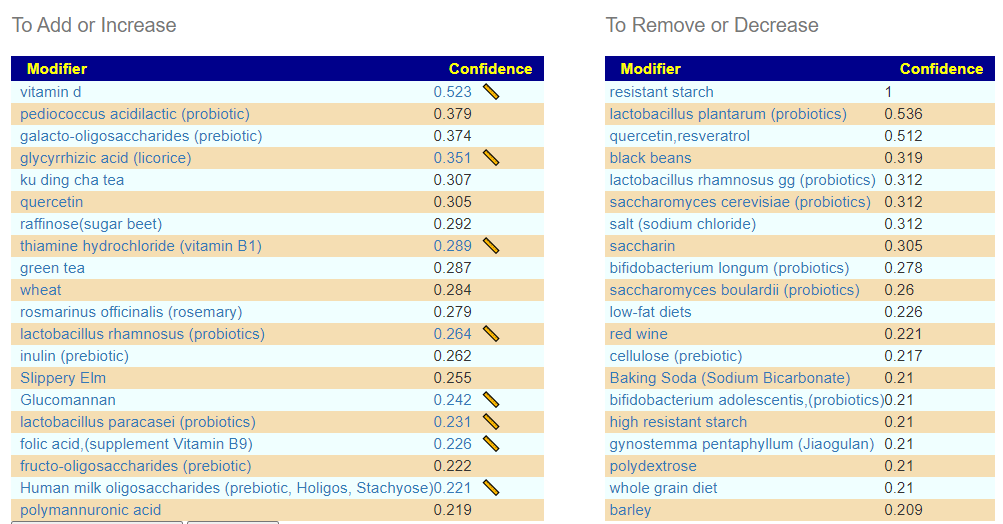
We have an idea of what may happen, apart from being a visual warning in the above diagrams to people considering FMT. The key question for this person is how to undo it. For this troublesome strain, we know some things that will increase or decrease it. I would focus on this strain alone and do a deep “spring cleaning” of supplements.
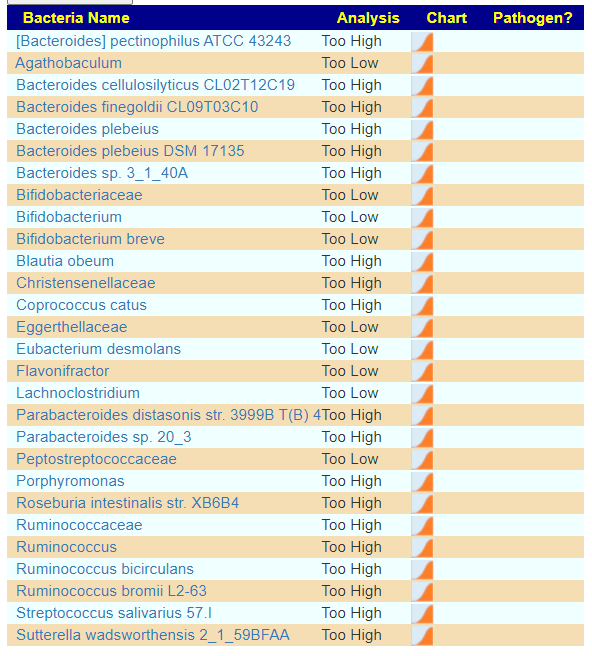
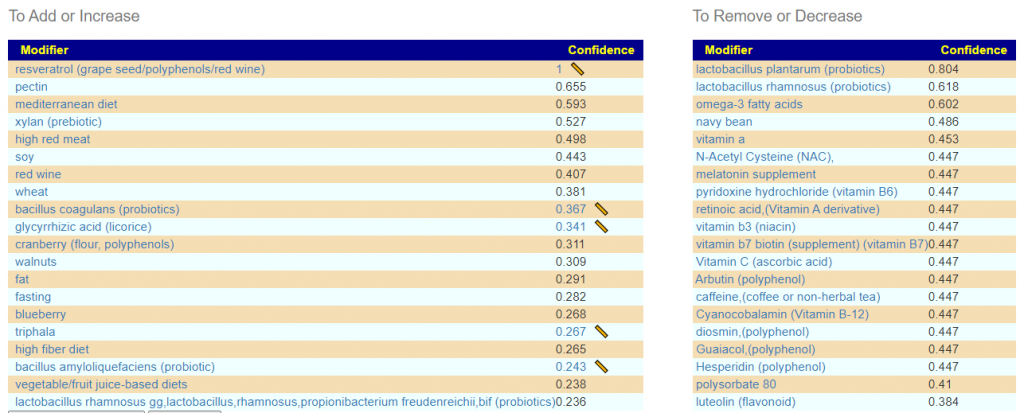
Looking at the Kaltoft-Moltrup Range based suggestions, we see a lot of bacteria selected as abnormal
On AFTER sanoke
And we see most of the above items are on the suggestions for this combination:
Bottom Line
I would suggest (after reviewing with your medical professional) the above changes and then do another Nirvana sample after 6-12 weeks to see what has changed. I would expect this Ruminococcus bicirculans. to be significantly reduced, the question is what will replace the 44% of the microbiome it currently occupies.
I was curious if we could infer something about the donor by looking at these changes. I looked at life style that could impact Ruminococcus bicirculans. etc and found that
A common sense (A priori) definition of what would be a healthy donor will often be someone that fits these two features, i.e. an athlete. ” Recent trends show more athletes trying a low-carbohydrate, high-fat (LCHF) diet for endurance performance. ” [Low vs. High Carbohydrate Diets for Endurance Performance] IMHO, this a priori common sense healthy donor is wrong — athletes have abnormal microbiomes. A priori is defined as “denoting reasoning or knowledge which proceeds from theoretical deductionrather than from observation or experience.“
The compatibility issue is not only at the microbiome but diet and exercise style. If the recipient does not consume the same diet as the donor, the transfer may go in odd directions. Similarly, we know exercise impacts the microbiome. A “desk jockey” getting the microbiome of someone that cycles for 4 hours a day will likely fade quickly.
Prior Posts on FMT
My interest in trying to understand different FMT responses go back at least 5 years. The following posts are likely worth reviewing. My feeling continues to be that the downside risk for the upside benefit is still too low to be a desirable course of action. Typically, it is an improvement that does not persist for extended periods. I believe a committed microbiome manipulation with regular retests and adjustment has considerably less downside and equivalent upside benefit that will likely persist longer — especially, if at least two samples and adjustments are done a year once sufficient benefit has been obtained.
I am tossing some technical information for those that are interested in how FMT can be done. There are many ways, including “the turkey baster” aka “Turd Burglars“. IMHO, it should be done under medical supervision with adequate testing (including comparing the donor’s microbiome, and life style to the recipient)
By Enema
This was used by the above
FMT with Enema – 6 doses, 1 every other day (11 days)
No antibiotics are used!
Pre- and probiotics (phgg, bifido)
Prior
Do not stop medication
follow a liquid diet the night before the first administration (and not the other 5!!!)
cleansing water enema in the morning before FMT
place the dose from the freezer in the refrigerator the night before. allow the dose to reach room temperature in the morning.
swallow 1 imodium after awakening (only) for the first dose
limit yourself to a light (liquid) meal
During
do not eat or drink anything during and immediately after the enema.
work in stages with a limited dose (but within 10 minutes on the left side)
use gravity to keep everything inside, e.g. a pillow under the hip
take a few deep breaths regularly
alternate position (left side, back, stomach, right side), every ten minutes (left is best for keeping inside)
regularly massage the dose upwards gently
try to keep the dose for ideally more than 6 hours
rest, relaxation and sleep are the best activities during the therapy days
After
do not immediately make major changes to your daily diet
not drinking and being (somewhat) thirsty helps to withdraw fluid from the injected dose
fibers help the new bacteria to settle. gradually introduce fiber into your diet. fiber rather through food than supplements.
” Dr Borody has overseen over 12,000 FMTs, creating a wealth of proprietary clinical data and insights.” Antibiotics are frequently used prior to FMT. He is likely the leading Australian MD that does FMT.
“Mild and transient side effects reported with the procedure include abdominal cramping, dis-comfort and bloating, belching, diarrhea, constipation, nausea and flatulence, which can occur while the infused bacteria are establishing themselves. A case of worsening inflammatory bowel disease, two cases of norovirus gastroenteritis and one case of E. coli bacteremia have also been reported, however FMT could not be established as the cause of these events” — NOTE The defensive “could not be established as the cause of these events” interpretation
Recently I revisited finding association between bacteria. We know bacteria both produce and consume metabolites and chemicals, as well as bacteriocins that will inhibit other bacteria. “Bacteriocins are potential alternatives to traditional antibiotics. These peptides, which are produced by many bacteria, can have a high potency and a low toxicity” {Nature 2012]. Finding the relationships has been a challenge because of the nature of the distribution (not a bell curve — see this post on the solution that I use for identifying abnormal values) Post #1Post #2.
This is a technical note (WARNING: Geek Speak) on the 262,603 relationship with correlation coefficient R2 of 0.10 or higher that is available on the site.
Because they have some shared ancestry, you would usually expect them to be friendly and suppurative of each other. The standard analysis is shown below, charting the counts from samples that have both bacteria.
Classic Approach
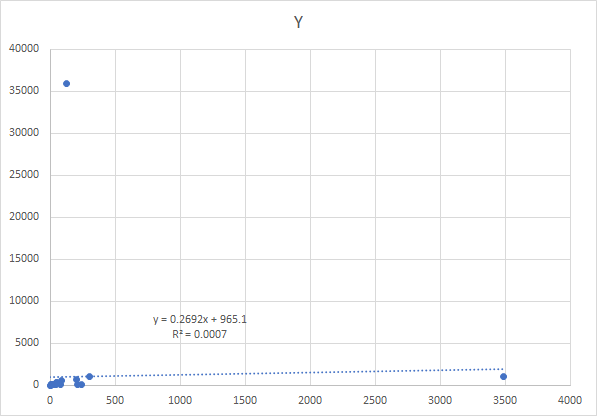
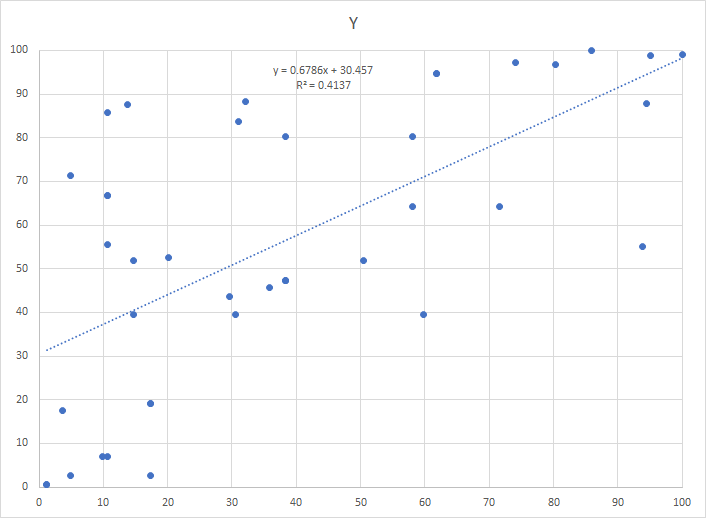
After an intro course to statistics, most people would do a regression. It is unlikely they would look at the chart because there are 2,669,956 charts that would be produced with the dataset that I am working with.
The regression and the chart is shown below, logical conclusion – no relationship.
The alternative is to use what is called monotonic increasing functions on the counts. We scale the function so that it’s range is 0 to 100. This preserves the nature of the data and discard the naïve assumption of linearity. The result is shown below. With this approach, we get the following chart. same data!!!
Each axis is the transformed count of a specific bacteria (Brucellaceae and Caulobacteraceae) from the same sample.
We could for each pair of bacteria derive the absolute optimal monotonic functions. This approach I find problematic because your appear to be fiddling with the data too much. I have put the additional constraint that you are allowed only one monotonic function per bacteria. I believe this will inhibit over-fitting the data to the model.
How many relationships over 0.1?
We have 1621 bacteria with at least one, and the top ones are shown below
taxonomy rank
taxonomy name
Count
family
Halanaerobiaceae
546
class
Fibrobacteria
526
class
Dehalococcoidia
506
family
Fibrobacteraceae
505
order
Fibrobacterales
501
genus
Fibrobacter
483
family
Nitrosomonadaceae
474
genus
Dehalogenimonas
474
order
Acidobacteriales
467
family
Micromonosporaceae
461
genus
Nitrosomonas
460
genus
Acinetobacter
459
family
Colwelliaceae
455
family
Acidobacteriaceae
453
What benefit does this give?
The impact of one bacteria on the other may be computed as slope * r2 . So R2 of .5 and a slope of .4 = .5 * .4 = .20 or 20%, thus for every 10 steps of one, the other will increase by 2.
We can use this when some bacteria X is high or low and we have no information on modifying it. We can look at the related bacteria with highest impact and its modifiers. We are trying to cascade by changing the associated bacteria to change our target bacteria! We are attempting to model the modifiers secondary changes into our suggestions.
Where is this on the site?
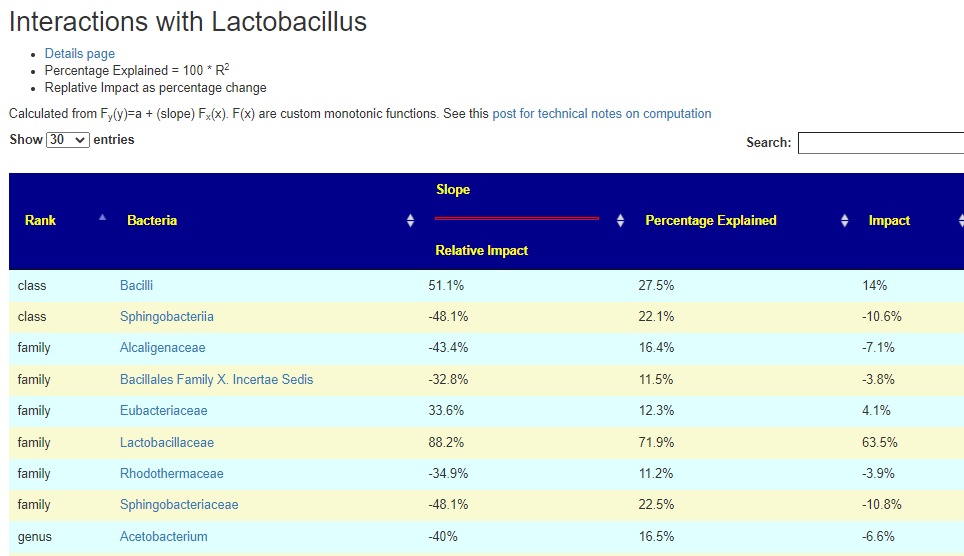
On the bacteria details pages. if there are associations, there will appear a link to it
Clicking this will take you to the impact page. In the example below you see that Lactobacillus accounts for 63% of it’s parent class. Lactobacillaceae(family) which includes Lactobacillus , Pediococcus , and Sharpea. So it is the greatest contributor the three.
Orphan Detail Pages
I call these orphan because there is not literature on them or little studies. For example Pectinatus where there was just one know citation, ginko. We now have 10 more marked with the association icon as shown below.
Available to include for Suggestions
There is a new checkbox on the custom suggestion page. If you wish these to be factored into suggestions just check the box.
A reader asked for a review. The reader had a prior sample taken 6 weeks before and specific treatments between
I took Ivermectin for 4 days during this month – one per day 12mg. I am treating yeast with nystatin 5,000 units 2 times per day for one month. I hope the die-off may have made room for bacteria to grow. I still feel crappy. I stopped lactobacillus ( I tried Lactobaciullis grains from Keith during that time. too much bloating), and started Akkermansia muciniphila and very recently some Bifidobacterium. I had cut back on dairy earlier (using soy milk and oat milk instead). I also took Sporonax (Itraconazole) antifungal about 5 times and Valtrex 500 mg 8 times.
My starting point (before looking at the samples) is to look at what we know about the impact of these items.
So the expectation of making room for bacteria appears very reasonable. The unfortunate aspect is that among the causalities are: Lactococcus, Lactobacillus, Bifidobacterium and Akkermansia muciniphila. So the question arises, will the good or the bad grow back faster?
What changed?
I first checked the common bacteria that most people are usually concerned with (cited above) and then will look at what increased. One item is of definite concern 💥, (class) Fusobacteria
(family) Fusobacteriaceae is listed as a should decrease, but it increased — it should be noted that this was very high in the prior one and more resistant and thus increased when the vacuum was created,
It was interesting to note the many of the bacteria that were abnormally high (95%ile) stayed the same or increased. The fact that they were high implies more aggressive strains (and possibly more bacteriocin and antibiotic resistant).
Looking at species that are outside of the KM range, we have the following being excessively high
Note that other Blautia species were abnormally low. These are already accounted for in the suggestions.
Looking further back
“I believe vancomycin started the problem to flare when I took in a couple of years ago. I also had my first covid shot of June 26, 2021 with immediate bad reactions and probably made worse since.
The very high Fusobacteria identified above is one bacteria that would be decreased by this antibiotic (so this is unlikely that this the cause of this being high).
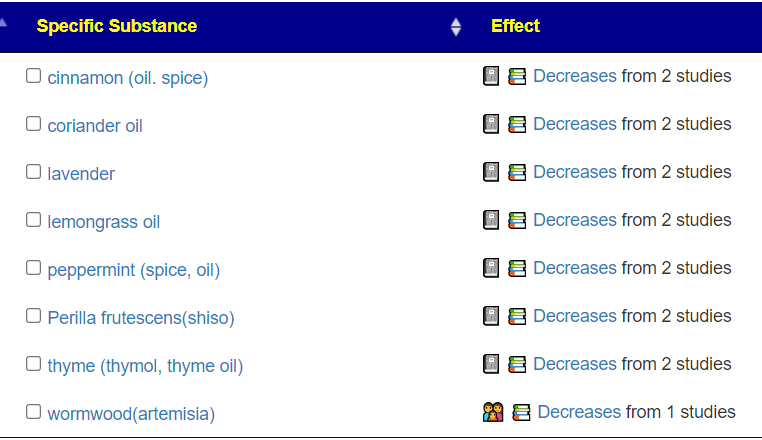
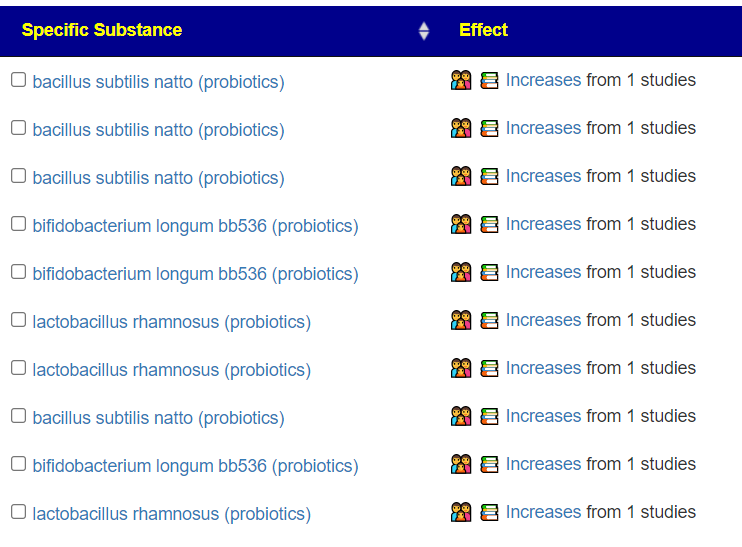
The following are items that have been reported to increase this bacteria:
I expect the suggestions to be very similar because the items high before stayed high (i.e. no change)
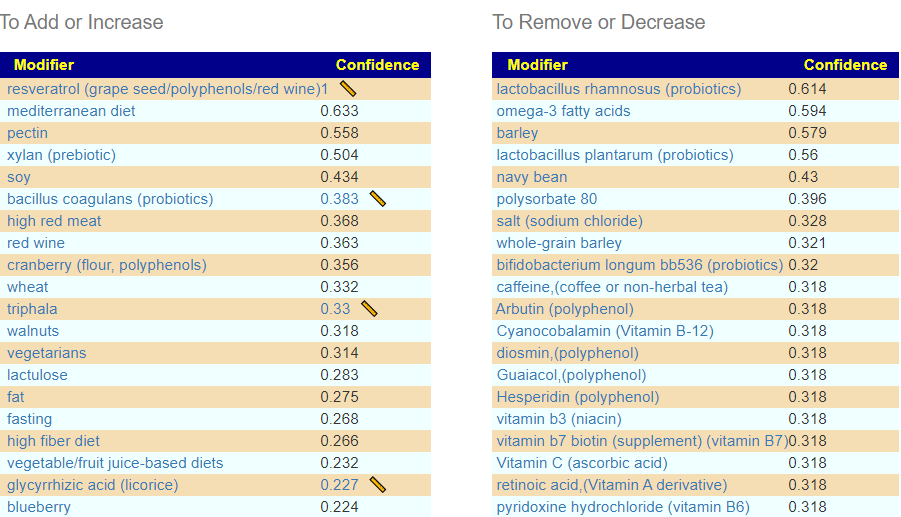
Suggestions were done with the Kaltoft-Moltrup ranges.Latest SuggestionsSuggestions from earlier sample
There was one additional probiotic suggested from the latest sample and it had a far stronger impact then the older ones.
Last SampleLPrior Sample
The KEGG suggested probiotics are the same ones, just a higher value because of an increased deficiency in enzymes being produced by the microbiome.
Latest SamplePrior Sample
Bottom Line
The data base correctly predicted the likely decrease of the common bacteria assumed important for health. Those that did no reduce were either not effected, more resistant strains (inferred from being high numbers prior)
What I found interesting was the absence of most probiotics (lactobacillus and bifidobacterium) in the safe suggestions, except for bifidobacterium breve. Securil (Propionibacterium freudenreichii) probiotic came in very strong.
Giloteaux65 found that supplementation with the bifidogenic substance Propionibacterium freudenreichii improved butyrate levels, which induces an anti-inflammatory cascade [2017]
One of particular interest is found in the product Securil and is called – Propionibacterium freudenreichii, which produce propionic acid, a natural biological acid that benefits the bifidus flora. [2010]
Note that this is specific for this person. Suggestions will be different for other peoples taking the same items.
Reader’s Desire
“I was also trying to get my methylation working better and taking b vitamins again. Could it be the b vitamins?”
The B-vitamins are suspect for the high levels of Fusobacteria. We should also note that are the to avoid list we see many of the B vitamins which suggests that much of the dysbiosis may be vitamin B related.
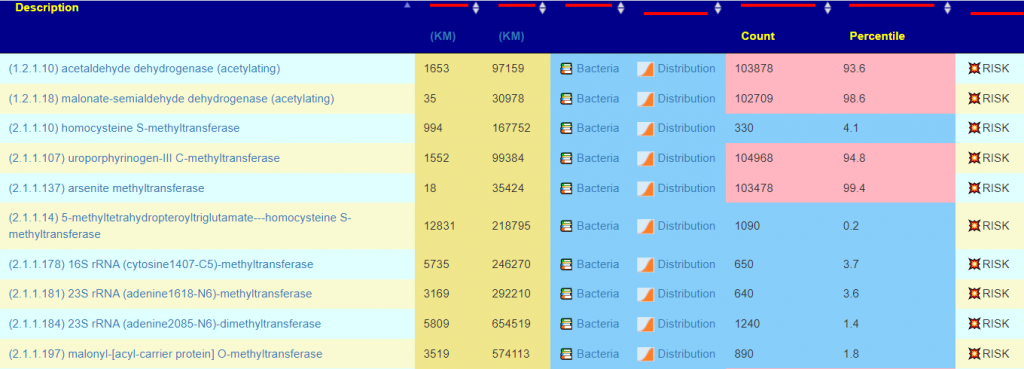
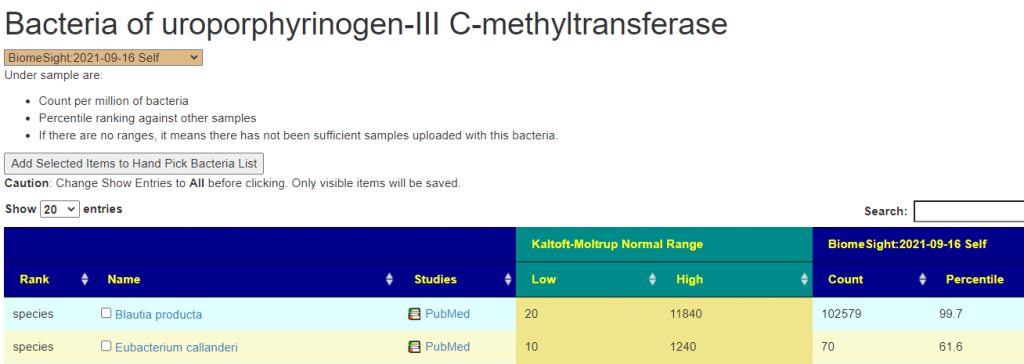
Concerning methylation, I usually see what enzymes are involved by checking with the Kyoto Encyclopedia of Genes and Genomes, And then check the KEGG Enzyme Outliers report. This report had a very high 470 items listed!!
A few of outliers showing extreme values
Drilling down into a few, we see some of the causes
This value is beyond the K-M range and thus suggestions to correct this is automatically captured in the above suggestions.
After a recent hospital visit for cellulitis (with many different antibiotics, both orally and by IV), my blood pressure was significantly elevated that the substitute MD (my usual was on vacation), that I was put on Lisinopril. Within a week I developed a dry cough that has for 35 years has been a “tell” for a relapse into Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Checking the literature, I found that about 30% of people develop this cough. To me it is an important tell, if it shows up — I need to do quick re-examination of what is going on. For a prescription drug to do so, really made me uncomfortable.
I then check Lisinopril against the bacteria shifts reported for ME/CFS, and it made them worst. In short, staying on it may well increase the risk of relapsing into ME/CFS. That is not acceptable.
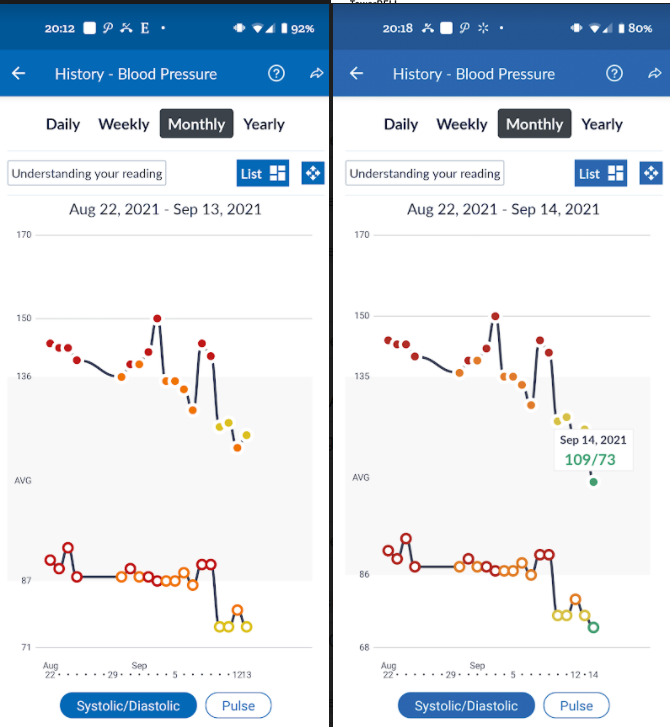
I stopped the lisinopril and proceeded to take the nutrients etc cited above, at or above the specified dosages. I know that it will take a little time for the microbiome to respond, but it did.
The spikes were from not waiting long enough after exercising. I now rest at least 20 minutes
Bifidobacterium Correlation
Reviewing the literature, there is the appearance of blood pressure being strongly associated with the amount of bifidobacterium as we age. Children are very high in Bifidobacterium and low in BP. As the typical amount of bifidobacterium decreases with age, blood pressure increases.
As a result, I add 2 tablespoons of bran to the typical 4 table spoons of oats porridge that was doing. I also added a package of Holigos (Human Milk Oligosaccharides) which I know is a super feeder of bifidobacterium.
This corresponded to the severe drop shown above.
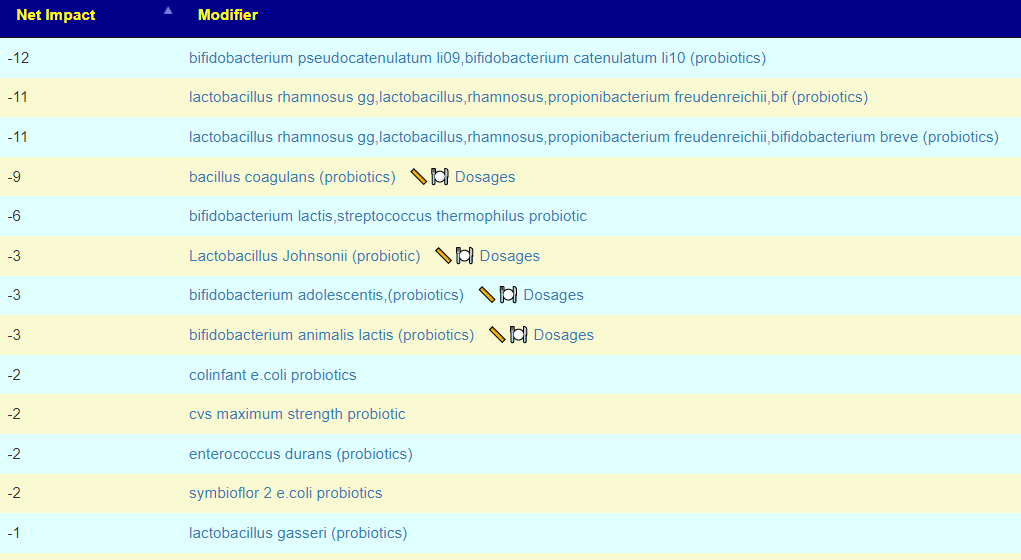
Possible Probiotic Impact
I was taking the following based on modelling of the bacteria shifts seen in hypertension:
Just got notification from the lab that last weeks was received.
One addendum, when I was in hospital for cellulitis, my potassium was very low and I required a (painful) IV of potassium. I examined the amount of potassium that my usual diet provided… It was very low, so I started to supplement with potassium citrate also.
I was able to normalize (for an almost 70 year old) blood pressure by using existing research and having patience. I believe the key items was encouraging bifidobacterium growth (sorry, bifidobacterium probiotics do not persist usually and have little impact), correcting mineral content (potassium, magnesium, calcium).
One more addendum, I usually did 10,000 steps a day with weekend hikes often being as high as 20,000 steps.
In an earlier post I cited supplements demonstrated in human clinical trials to lower blood pressure. A still earlier post looked at gut bacteria associated with hypertension and hypotension. I have collected the bacteria shifts reported from studies published on PubMed here
Literature of the microbiome bacteria
The following are the sources for the bacteria information
Administration with Quinoa Protein Reduces the Blood Pressure in Spontaneously Hypertensive Rats and Modifies the Fecal Microbiota. Nutrients (Nutrients ) Vol: 13 Issue 7 Pages: Pub: 2021 Jul 17 Epub: 2021 Jul 17 Authors Guo H , Hao Y , Fan X , Richel A , Everaert N , Yang X , Ren G , SummaryHtml ArticlePublication
Changes of gut microbiome composition and metabolites associated with hypertensive heart failure rats. BMC microbiology (BMC Microbiol ) Vol: 21 Issue 1 Pages: 141 Pub: 2021 May 5 Epub: 2021 May 5 Authors Li L , Zhong SJ , Hu SY , Cheng B , Qiu H , Hu ZX , SummaryHtml ArticlePublication
Improvement of intestinal flora: accompany with the antihypertensive effect of electroacupuncture on stage 1 hypertension. Chinese medicine (Chin Med ) Vol: 16 Issue 1 Pages: 7 Pub: 2021 Jan 7 Epub: 2021 Jan 7 Authors Wang JM , Yang MX , Wu QF , Chen J , Deng SF , Chen L , Wei DN , Liang FR , SummaryHtml ArticlePublication
Associations between gut microbiota, faecal short-chain fatty acids, and blood pressure across ethnic groups: the HELIUS study. European heart journal (Eur Heart J ) Vol: 41 Issue 44 Pages: 4259-4267 Pub: 2020 Nov 21 Epub: Authors Verhaar BJH , Collard D , Prodan A , Levels JHM , Zwinderman AH , Bäckhed F , Vogt L , Peters MJL , Muller M , Nieuwdorp M , van den Born BH , SummaryHtml ArticlePublication
Changes in the Gut Microbiota are Associated with Hypertension, Hyperlipidemia, and Type 2 Diabetes Mellitus in Japanese Subjects. Nutrients (Nutrients ) Vol: 12 Issue 10 Pages: Pub: 2020 Sep 30 Epub: 2020 Sep 30 Authors Takagi T , Naito Y , Kashiwagi S , Uchiyama K , Mizushima K , Kamada K , Ishikawa T , Inoue R , Okuda K , Tsujimoto Y , Ohnogi H , Itoh Y , SummaryHtml ArticlePublication
Human genetic determinants of the gut microbiome and their associations with health and disease: a phenome-wide association study. Scientific reports (Sci Rep ) Vol: 10 Issue 1 Pages: 14771 Pub: 2020 Sep 8 Epub: 2020 Sep 8 Authors Groot HE , van de Vegte YJ , Verweij N , Lipsic E , Karper JC , van der Harst P , SummaryHtml ArticlePublication
Differential Analysis of Hypertension-Associated Intestinal Microbiota. International journal of medical sciences (Int J Med Sci ) Vol: 16 Issue 6 Pages: 872-881 Pub: 2019 Epub: 2019 Jun 2 Authors Dan X , Mushi Z , Baili W , Han L , Enqi W , Huanhu Z , Shuchun L , SummaryHtml ArticlePublication
Critical Role of the Interaction Gut Microbiota – Sympathetic Nervous System in the Regulation of Blood Pressure. Frontiers in physiology (Front Physiol ) Vol: 10 Issue Pages: 231 Pub: 2019 Epub: 2019 Mar 8 Authors Toral M , Robles-Vera I , de la Visitación N , Romero M , Yang T , Sánchez M , Gómez-Guzmán M , Jiménez R , Raizada MK , Duarte J , SummaryHtml ArticlePublication
DIFFERENCES IN MICROBIOME IN RAT MODELS OF CARDIOVASCULAR DISEASE. South African journal of surgery. Suid-Afrikaanse tydskrif vir chirurgie (S Afr J Surg ) Vol: 55 Issue 2 Pages: 71 Pub: 2017 Jun Epub: Authors Thiba A , Umar CA , Myende S , Nweke E , Rumbold K , Candy G , Summary
Altered Gut Microbiome Profile in Patients With Pulmonary Arterial Hypertension. Hypertension (Dallas, Tex. : 1979) (Hypertension ) Vol: Issue Pages: HYPERTENSIONAHA11914294 Pub: 2020 Feb 24 Epub: 2020 Feb 24 Authors Kim S , Rigatto K , Gazzana MB , Knorst MM , Richards EM , Pepine CJ , Raizada MK , SummaryPublicationPublication
Intestinal Flora Modulates Blood Pressure by Regulating the Synthesis of Intestinal-Derived Corticosterone in High Salt-Induced Hypertension. Circulation research (Circ Res ) Vol: Issue Pages: Pub: 2020 Feb 13 Epub: 2020 Feb 13 Authors Yan X , Jin J , Su X , Yin X , Gao J , Wang X , Zhang S , Bu P , Wang M , Zhang Y , Wang Z , Zhang Q , SummaryPublicationPublication
Exercise and food supplement of vitamin C ameliorate hypertension through improvement of gut microflora in the spontaneously hypertensive rats. Life sciences (Life Sci ) Vol: 269 Issue Pages: 119097 Pub: 2021 Mar 15 Epub: 2021 Jan 19 Authors Li Y , Zafar S , Salih Ibrahim RM , Chi HL , Xiao T , Xia WJ , Li HB , Kang YM , SummaryPublication
Enterococcus faecalis contributes to hypertension and renal injury in Sprague-Dawley rats by disturbing lipid metabolism. Journal of hypertension (J Hypertens ) Vol: 39 Issue 6 Pages: 1112-1124 Pub: 2021 Jun 1 Epub: Authors Zhu Y , Liu Y , Wu C , Li H , Du H , Yu H , Huang C , Chen Y , Wang W , Zhu Q , Wang L , SummaryPublication
Bifidobacterium reduction is associated with high blood pressure in children with type 1 diabetes mellitus. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie (Biomed Pharmacother ) Vol: 140 Issue Pages: 111736 Pub: 2021 Aug Epub: 2021 May 23 Authors Lakshmanan AP , Shatat IF , Zaidan S , Jacob S , Bangarusamy DK , Al-Abduljabbar S , Al-Khalaf F , Petroviski G , Terranegra A , SummaryPublication
Gut microbiome diversity and composition is associated with hypertension in women. Journal of hypertension (J Hypertens ) Vol: Issue Pages: Pub: 2021 May 10 Epub: 2021 May 10 Authors Louca P , Nogal A , Wells PM , Asnicar F , Wolf J , Steves CJ , Spector TD , Segata N , Berry SE , Valdes AM , Menni C , SummaryPublication
Probitotics
From Theoretical Model
These are based on the bacteria reported above and an AI engine on the impact of various probiotics (in order of confidence). Links to dosages found to be sufficient to cause changes in clinical trails are after each. It is a common mistake to take ‘homeopathic” dosage, that is, if a product contains one of these, then that is sufficient. This thinking is akin to thinking that a squirt gun is sufficient to put out a wildfire!
” The fermentation of beans with Bacillus Subtilis B060 may therefore constitute a successful strategy for producing a functional food with antihypertensive activity.” [2014]
“water extracts of Bacillus subtilis-fermented pigeon pea (100 mg/kg body weight) significantly improved systolic blood pressure (21 mmHg) and diastolic blood pressure (30 mmHg) in spontaneously hypertensive rats.” [2015]
” treatment of model rats with Lactobacillus rhamnosus GG prevented aggravation of hypertension by reducing blood TMAO levels, modulating Th1/Th2 cytokine imbalance and suppressing phosphorylation levels of ERK1/2, Akt and mTOR.”[2019]
From Human Studies
NO EFFECT from Lactobacillus plantarum PS128 [2031] -dosage was sufficient
“The clinical significance of blood pressure-lowering effect of Lactobacillus Plantarum supplementation is not considerable; ” [2020]
“Our meta-analysis showed a modest but a significant reduction in SBP and DBP in patients with hypertension, particularly in those with diabetes mellitus, following probiotic supplementation. This effect was associated with treatment duration, dosage, and the age of subject but was not associated with single or multiple strains usage. Additionally, probiotic supplement had a beneficial effect in reducing BMI and blood glucose.” [2020]
“Lactobacillus consumption significantly reduced systolic blood pressure (SBP) by -2.74 mmHg and diastolic blood pressure (DBP) by -1.50 mmHg when comparing with the control group.” Dosage above 5 Billion CFU per day. [2020]
“Probiotic consumption significantly changed systolic BP by -3.56 mm Hg and diastolic BP by -2.38 mm Hg compared with control groups.” After 8 weeks at 10+ Billion CFU/day [2014] No strains or species specified
“Lactobacillus para casei LPC-37, Lactobacillus rhamnosus HN001, Lactobacillus acidophilus NCFM, and Bifidobacterium lactis HN019 (109 CFU of each strain) for 8 weeks….Probiotic supplementation lowered, although without statistical significance, systolic BP by about 5 mmHg and diastolic BP by about 2 mmHg in hypertensive women.” [2020]
Bacillus subtilis (especially the Natto strain) appears to be most effective, both from a theoretical and animal study point of view. The theoretical model appears to work reasonably. Probiotic consumption appears to do no harm from clinical studies — however, the theoretical model indicates some may increase the microbiome shifts in the wrong direction.
This post is an update of an earlier post. It deals mainly with non-prescription items. Some prescription items can have adverse effects on the microbiome seen with other conditions. “No medical condition is an Island“
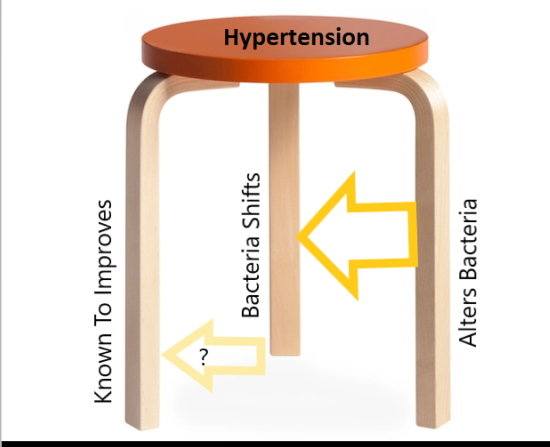
After reviewing reviewed tested supplements, we use the Three-Legged-Stool model to get additional candidates and then check if there are studies supporting their use.
Prescription Responder and Non-Responders
I came across this 2021 article that was investigating DNA/SNP and hypertension drugs.
“Drug effectiveness was defined as 10% decrease in systolic blood pressure at 1 week follow-up. “
For modelling substances for a condition, I often use a three legged tool as shown below
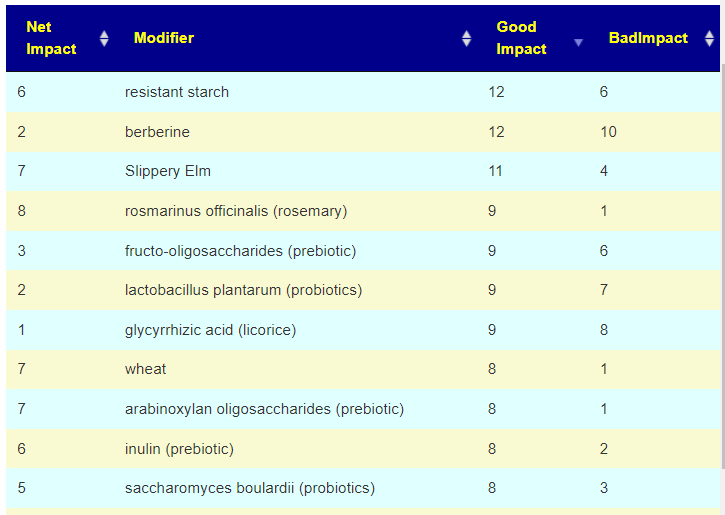
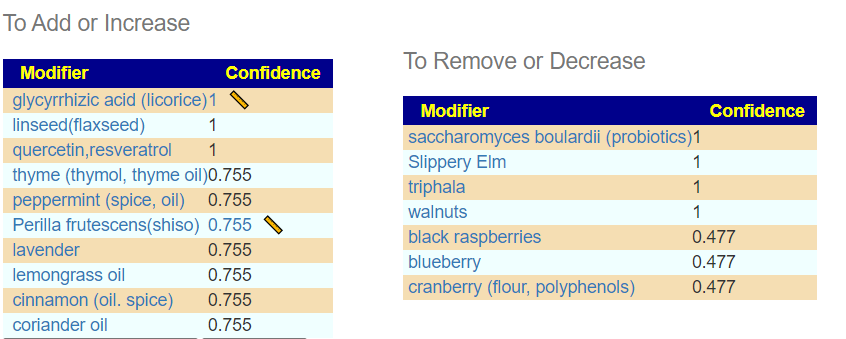
Items were ranked by number of bacteria favorability impacted. The top 3 suggested modifiers are below. The next step is to see if there is any literature. [Good Impact: Bad Impact]
Effect of Lactobacillus plantarum containing probiotics on blood pressure: A systematic review and meta-analysis [2020]
“A statistically significant reduction in systolic blood pressure was also observed”[2017]
glycyrrhizic acid (licorice) [9:8}
“Women taking licorice have experienced elevated blood pressure” [2021]
” No electrolyte abnormality, significant changes in blood pressure or blood glucose levels were observed during the [Licorice] study.” [2019]
appear to cause hypertension in association with potassium chloride [2018]
rosmarinus officinalis (rosemary) [9:1]
“Both blood pressure variables of SBP and DBP reflect the clinically significant antihypotensive effect of Rosemary essential oil that was maintained throughout the treatment period. ” [2014]
fructo-oligosaccharides (prebiotic) [9:6]
Nothing
The next items
zinc [8:3]
“Angiotensin-converting enzyme (ACE) is a zinc-dependent dicarboxypeptidase ” [2021] – impacts prescription hypertension medicines
“Zinc and copper might be not independently associated with hypertension in US adults.” [2018]
saccharomyces boulardii (probiotics) [8:3]
nothing
wheat [8:1] – complex, ancient varieties appear to have benefits
“Antihypertensive and antioxidant activities of enzymatic wheat bran protein hydrolysates” [2020]
Short-Term Hemodynamic Effects of Modern Wheat Products Substitution in Diet with Ancient Wheat Products: A Cross-Over, Randomized Clinical Trial [2018] SBP decreased
arabinoxylan oligosaccharides (prebiotic) [8:1]
nothing
inulin (prebiotic) [8:2]
Inulin Supplementation Reduces Systolic Blood Pressure in Women with Breast Cancer Undergoing Neoadjuvant Chemotherapy [2019] SBP: -4 mm Hg
lactobacillus salivarius (probiotics) [7:1]
Nothing
vitamin a [7:2]
Inverse association between dietary vitamin A intake and new-onset hypertension[2021] “Our results emphasized the importance of maintaining relatively higher vitamin A intake levels for the prevention of hypertension.”
oregano (origanum vulgare, oil) |[7:2]
Nothing
We do see some items from our first list, with predictions tending to agree. Remember that we are doing a naïve count by bacteria and dealing with fuzzy data
This illustrates the use of the three legged stool approach for treating conditions. The use of microbiome appears to produce an extended list of candidates substances that appears to be in general agreement with studies. Each candidate substance should be researched because we have a complex mixture of bacteria.
A reader emails as shown below. The deep vein thrombosis (DVT) aspect interest me because it is typically associated with inherited coagulation defects (an interest that I have), although actual DVT has not been an interest (when I flew regularly, I was prescribed heparin and took it with piracetam (good for my specific defect).
On 15 July 2021 I uploaded my first Thryve sample to your website. Prior to that sample I had a few Ubiome samples from a few years ago. Thryve seems to detect many more bacteria than Ubiome. My results seem particularly unusual with numerous rare bacteria unfortunately. My main symptoms have always been constipation, food allergies and 2 episodes of DVT whereby I remain on anticoag therapy to avoid further recurrences. Bacteroides Vulgatus seems to be significantly high in all of my test results, both Ubiome and Thryve. I wonder whether this could be the ‘root cause’ given that the numbers of it are so much higher than any other bacteria.
What is known about DVT and Microbiome?
After a while searching PubMed, I finally found a 2020 article. Of special interest is Staphylococcus aureus which appears to have a significant role with ME/CFS [2016 Post] and may account for the high percentage of hypercoagulation seen there.
” Many known bacteria, such as Helicobacter pylori, Chlamydia pneumoniae, Mycoplasma pneumoniae, Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, and Escherichia coli, causing infections may increase the risk of thrombotic complications through platelet activation or may lead to an inflammatory reaction involving the fibrinolytic system.” Microbial Modulation of Coagulation Disorders in Venous Thromboembolism [2020]
A fuller list from the full article (Citing 2019) is, below
Staphylococcus aureus,
Streptococcus pyogenes
Pseudomonas aeruginosa,
Escherichia coli,
Klebsiella pneumoniae,
Chlamydia pneumoniae,
Helicobacter pylori,
Haemophilus influenzae
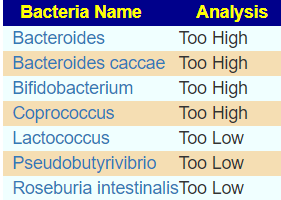
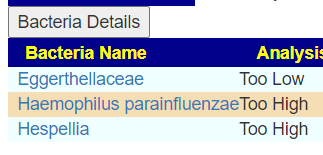
Looking at samples over three year, we have a strong candidate for causing DVT, Haemophilus parainfluenzae. It is consistently very high. Blank indicated no reported value.
Using the Nat.Library of Medicine filter for constipation and relaxing to include high and low 12%, we came up with only a very short list of candidates. Using Kaltoft-Møldrup bounds, nothing was selected.
One study found Haemophilus influenzae associated with GI issues [2004]
As for allergies, “Streptococcus pneumoniae, Haemophilus influenzae and Moraxella catarrhalis are often involved in respiratory infections associated with wheezing, but there is no evidence for their active role in asthma pathogenesis or exacerbation. ” [2009]
Running Advance Suggestions “as Is”
The key items selected reflects our analysis above:
KEGG Suggested Probiotics
The numbers are low, indicating no major issues. None of the suggestions are known to increase (or decrease) our focus. For supplements, it is similar
beta-alanine
NADH
Bottom Line
The primary question from the user appears to be answered. I would suggest fixing the Haemophilus parainfluenzae in isolation from the other two issues. Those two issues resolution will likely tend towards the use of probiotics — which are counter indicated with Haemophilus parainfluenzae. You have to prioritize issues and be careful not to send mix messages to the microbiome.
As always, review and consult with your medical professional before implementing
A reader sent me this email (actually another one did too). The issue was fixed but not in the obvious way:
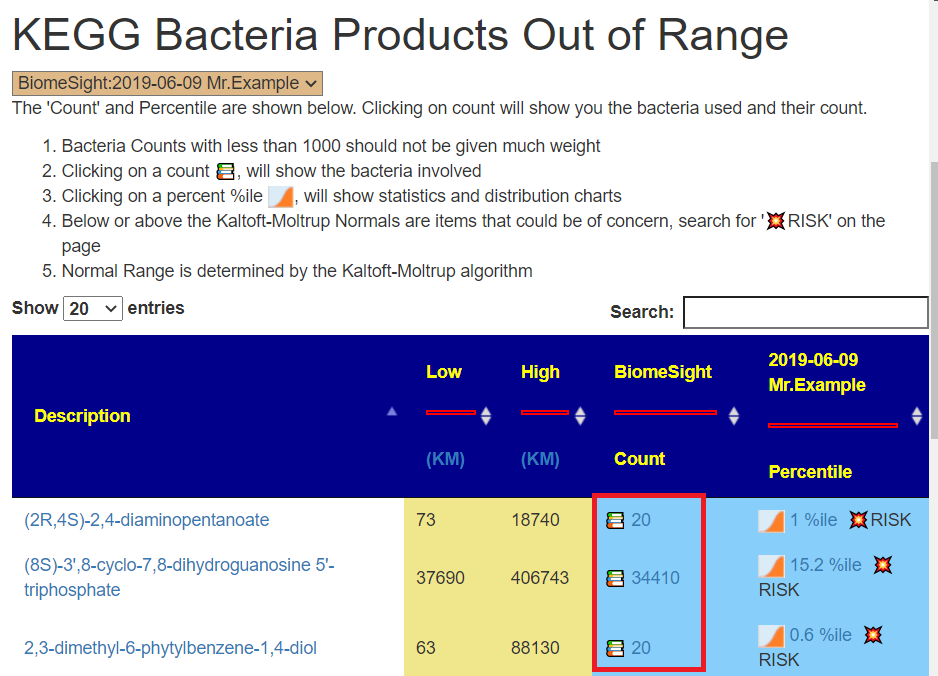
BUG: Checking and “Add elected items…”
This was actually a coding error, the checkbox should not be there. To do what the user intended:
Now to pick bacteria for KEGG Vectors
Below is the revised page fixing the problem. Notice the RED RECTANGLE
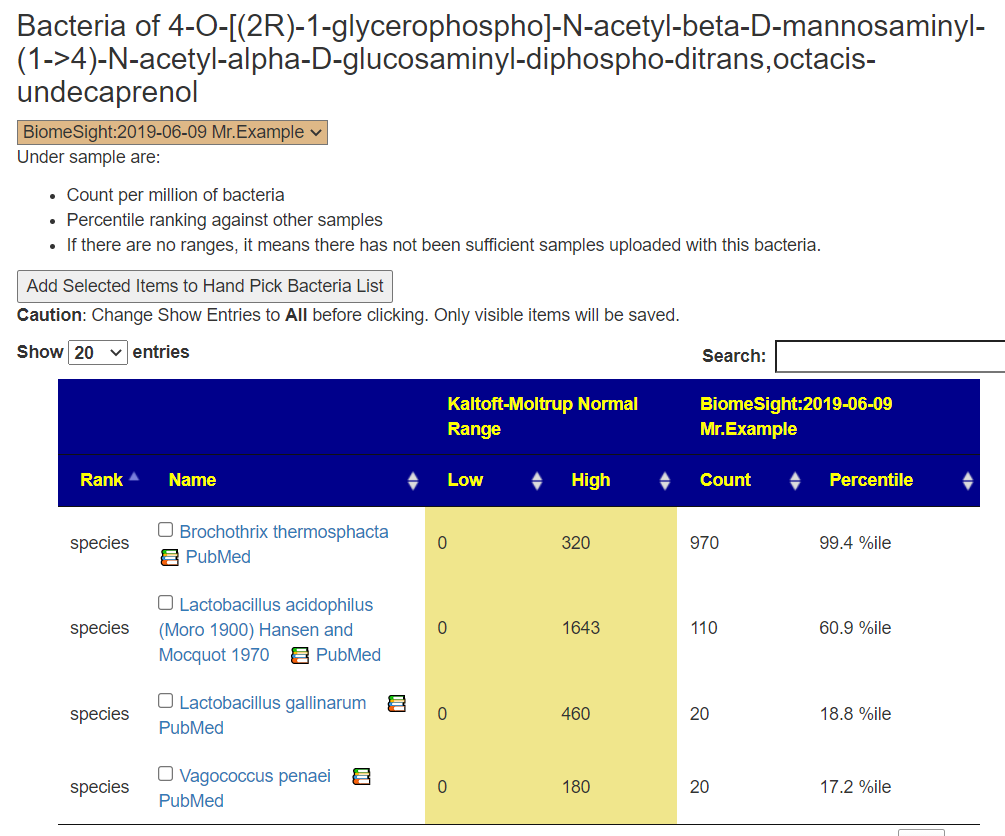
Clicking this will take you to the page listing the bacteria for this item, example below
On this page the checkboxes work. You see all of the bacteria associated that you have and thus can target the specific one (likely Brochothrix thermosphacta that is running well above the top of the normal range) that is causing the KEGG Vector (Product, Enzyme, Module) to be of concern.










Recent Comments