If something is wrong, then one of the following steps may have failed. See if you can identify which one and then do the suggested way to correct it.
Transferring Data from your Computer to the Database
- File is uploaded
- File is broken down into individual lines
- Each line is parse (broken apart). The following errors have occurred:
- The provider changed their file format. We show sample of what we expect to see on each upload page. If your file does not match, send the file to Research@MicrobiomePrescription.com so we may add a new adapter.
- The user read the file into Excel or some other program and then saved it. This can cause tabs to be replaced with commas and other format changes that breaks our adapter. Obtain a fresh copy from your provider and directly upload it.
- If the file does not contain NCBI taxon numbers, we proceed to do name matching
- Many bacteria have dozens of names, we do best efforts for matching
- We log names that we have failed to match for periodic review and addition to our lookup database (3,016,747 names at present)
- We convert percentages or other measures to counts out of one million (old ubiome standard)
Your Sample Has been Saved to the Database at this point
It is possible that some of the processing described below failed. If a table or chart appears wrong, You can do each step by hand to attempt to correct this failure to process by using the last drop down on the samples page.
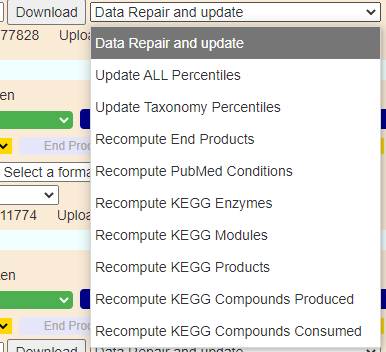
- We then do Upload PostProcessing which consists of the steps below [Note: the source data rarely changes for these – if they do, we do a bulk update of all samples]
- Computing a Hash of the upload (so we can identify duplicates – please delete any duplicates)
- Update our online bacteria database for any new bacteria not seen (at present we have 19,590 different bacteria[NCBI numbers] reported from different tests)
- We add missing members of the NCBI hierarchy
- Some labs do not report the complete hierarchy, or use their own.
- We apply NCBI rules for what is the parent of a particular bacteria.
- This allows a uniform presentation across many labs
- There have been odd conflicts — where the count of the children of a family exceeds what the lab reported for the family. The root cause is that they are using a different hierarchy. We keep the lab’s value but it can result in some odd Krona Charts and other visual presentations.
- We identify and count bacteria that are rarely seen in other samples (1%,2%,4%,8%,16%)
- We then compute some Health Statistics
- GHMI Healthy, UnHealthy
- Condition Health
- We compute KEGG Module raw numbers
- We compute KEGG Product raw numbers
- We compute KEGG Enzymes raw numbers
- We compute KEGG Substrate raw Products
- We compute KEGG Compound Produced
- We compute KEGG Compound Consumed (Substrate)
- We compute End Product raw numbers
- We now proceed to add percentiles to the above 6 tables(Bacteria Count, End Product, K-Module, K-Product, K-Enzymes, K-Substrate) using daily recomputed distribution tables:
- Taxonomy Reference
- End Product Reference
- K-Enzymes Reference
- K-Module Reference
- K-Product Reference
- K-Substrate Reference
- K-Compound Produced Reference
- K-Compound Consumed Reference
PubMed Medical Conditions Handling
Most studies on the US National Library of Medicine reports cohorts averages where above or below control averages with statistical significance. Averages are not the right measure, but it is what is being used…. We punt by using 75%ile and above for a match for higher, and 25% and below for lower.
After the above processing (so we have percentile to work from) we compute these numbers
Recent Comments